Regulation of Protein Post-Translational Modifications on Metabolism of Actinomycetes


Abstract
:1. Introduction
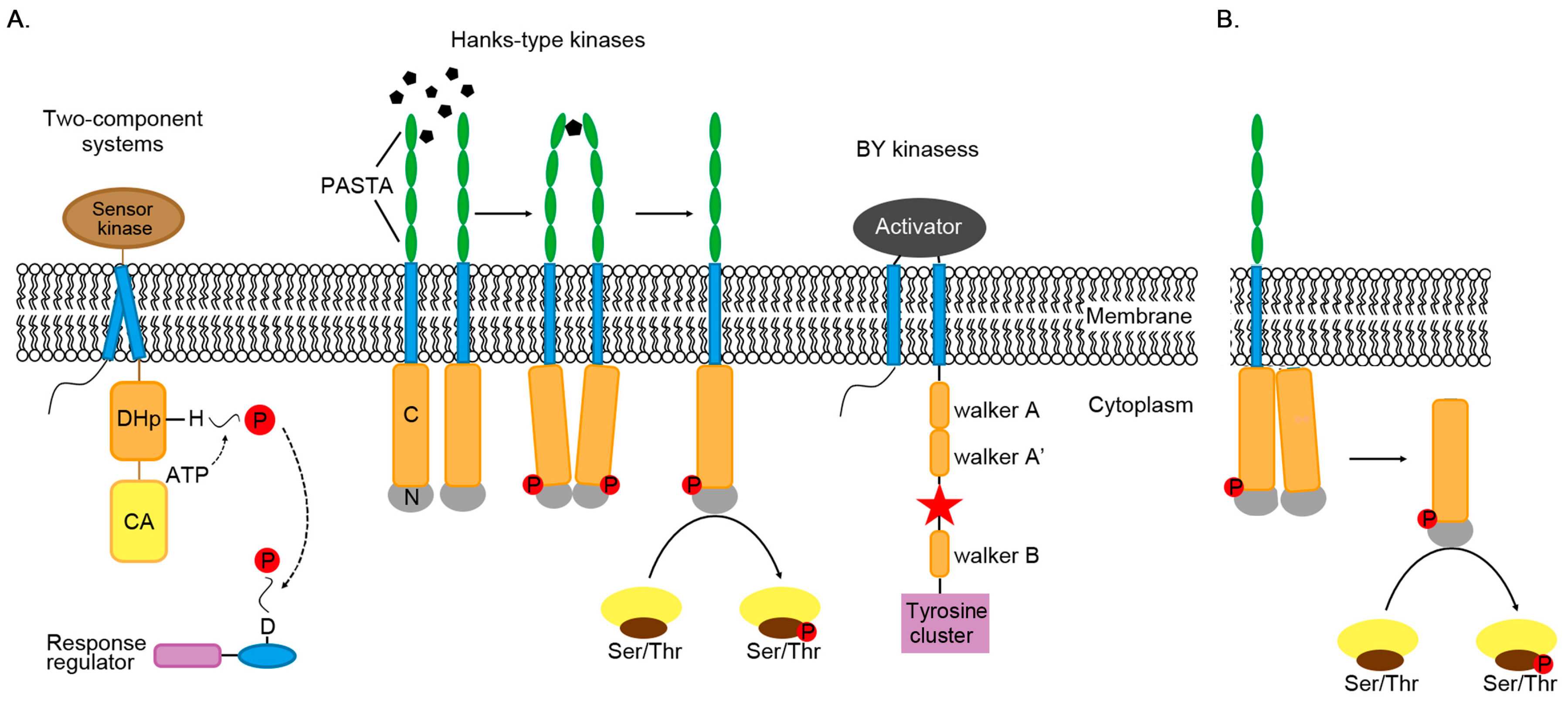
2. Phosphorylation
2.1. Two-Component Systems
2.2. Serine/Threonine/Tyrosine Phosphorylation
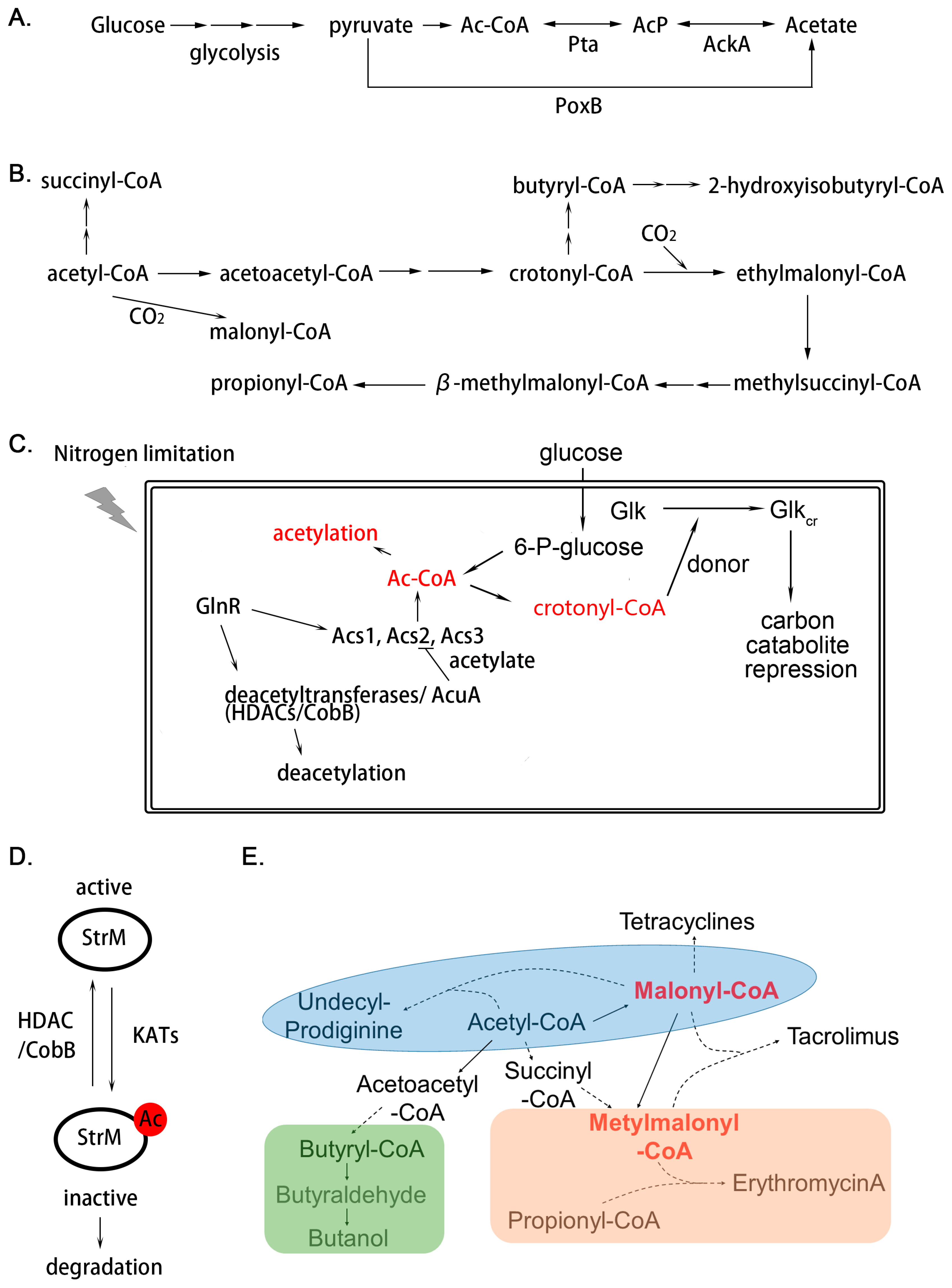
3. Acylation
3.1. How Acetylation and Deacetylation Occur
3.2. The Roles of Acetylation in Actinobacteria
3.2.1. Acetylation Functions in Cellular Signaling for Nutrient Assimilation
3.2.2. Acetylation Plays a Key Role in Secondary Metabolism
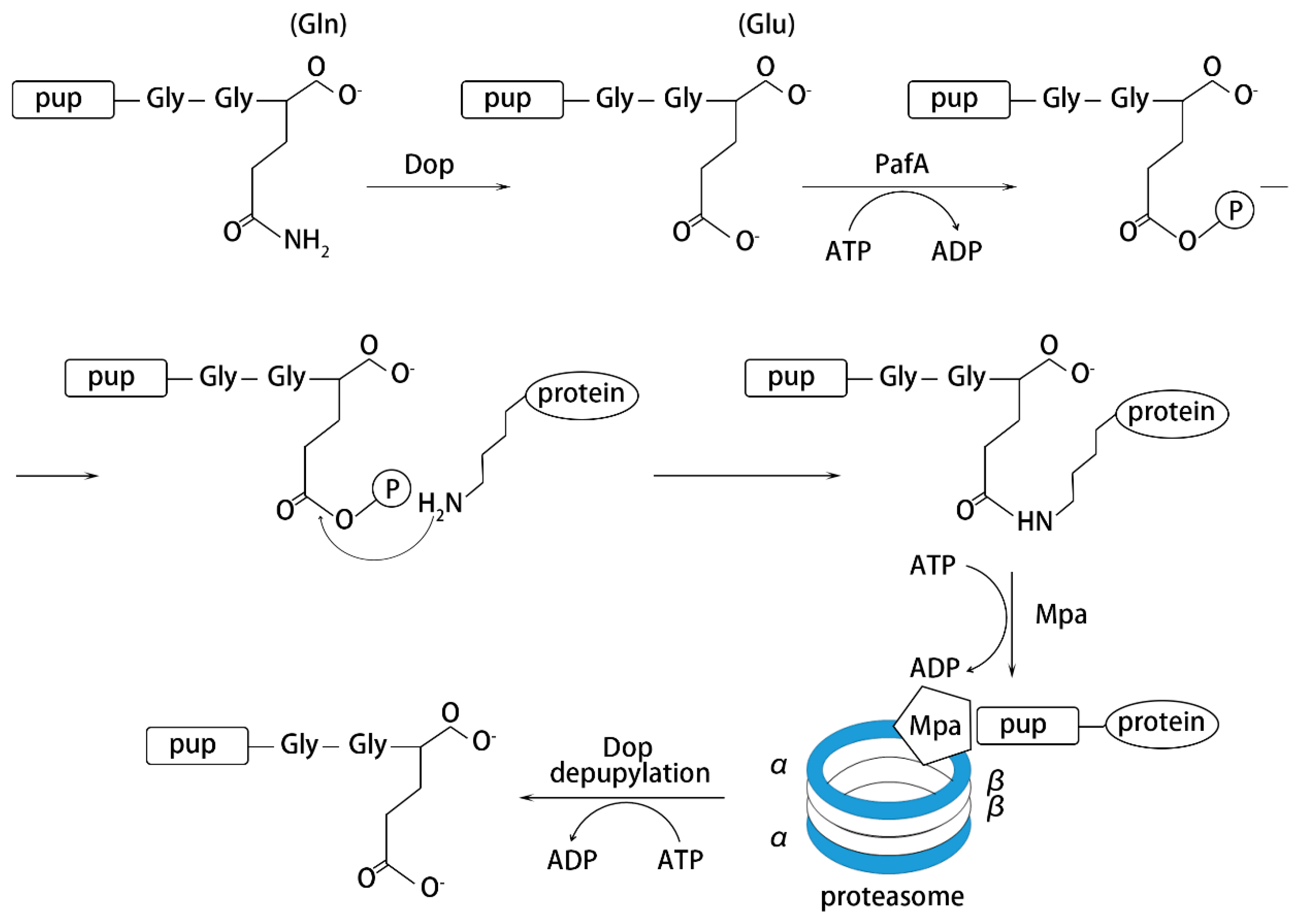
4. Pupylation
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Meier-Kolthoff, J.P.; Klenk, H.P.; Clement, C.; Ouhdouch, Y.; Van Wezel, G.P. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urem, M.; Swiatek-Polatynska, M.A.; Rigali, S.; Van Wezel, G.P. Intertwining nutrient-sensory networks and the control of antibiotic production in Streptomyces. Mol. Microbiol. 2016, 102, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutledge, P.J.; Challis, G.L. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 2015, 13, 509–523. [Google Scholar] [CrossRef]
- Martín, J.F.; Liras, P. Engineering of regulatory cascades and networks controlling antibiotic biosynthesis in Streptomyces. Curr. Opin. Microbiol. 2010, 13, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Van Wezel, G.P.; McDowall, K.J. The regulation of the secondary metabolism of Streptomyces: New links and experimental advances. Nat. Prod. Rep. 2011, 28, 1311–1333. [Google Scholar] [CrossRef] [PubMed]
- Romero-Rodriguez, A.; Robledo-Casados, I.; Sanchez, S. An overview on transcriptional regulators in Streptomyces. Biochim. Biophys. Acta 2015, 1849, 1017–1039. [Google Scholar] [CrossRef]
- Martin, J.F.; Sola-Landa, A.; Santos-Beneit, F.; Fernandez-Martinez, L.T.; Prieto, C.; Rodriguez-Garcia, A. Cross-talk of global nutritional regulators in the control of primary and secondary metabolism in Streptomyces. Microb. Biotechnol. 2011, 4, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Macek, B.; Forchhammer, K.; Hardouin, J.; Weber-Ban, E.; Grangeasse, C.; Mijakovic, I. Protein post-translational modifications in bacteria. Nat. Rev. Microbiol. 2019, 17, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, A.R.; Chandra, G.; Shaw, A.D.; Rowland, J.J.; Kell, D.B.; Bibb, M.J.; Chater, K.F. Primary and secondary metabolism, and post-translational protein modifications, as portrayed by proteomic analysis of Streptomyces coelicolor. Mol. Microbiol. 2002, 46, 16. [Google Scholar] [CrossRef] [Green Version]
- Ishigaki, Y.; Akanuma, G.; Yoshida, M.; Horinouchi, S.; Kosono, S.; Ohnishi, Y. Protein acetylation involved in streptomycin biosynthesis in Streptomyces griseus. J. Proteom. 2017, 155, 63–72. [Google Scholar] [CrossRef]
- Rioseras, B.; Shliaha, P.V.; Gorshkov, V.; Yague, P.; Lopez-Garcia, M.T.; Gonzalez-Quinonez, N.; Kovalchuk, S.; Rogowska-Wrzesinska, A.; Jensen, O.N.; Manteca, A. Quantitative proteome and phosphoproteome analyses of Streptomyces coelicolor reveal proteins and phosphoproteins modulating differentiation and secondary metabolism. Mol. Cell Proteom. 2018, 17, 1591–1611. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Sha, W.; Liu, Z.; Tang, T.; Liu, H.; Qin, L.; Cui, Z.; Chen, J.; Liu, F.; Zheng, R.; et al. Lysine acetylation of DosR regulates the hypoxia response of Mycobacterium tuberculosis. Emerg. Microbes Infect. 2018, 7, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Yang, W.; Fan, X.; Xie, J. Comprehensive analysis of protein acetyltransferases of human pathogen Mycobacterium tuberculosis. Biosci. Rep. 2019, 39, BSR20191661. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.F.; Xu, W.F.; Zhao, Q.W.; Luo, S.; Chen, X.A.; Li, Y.Q.; Mao, X.M. Crotonylation of key metabolic enzymes regulates carbon catabolite repression in Streptomyces Roseosporus. Commun. Biol. 2020, 3, 192. [Google Scholar] [CrossRef] [PubMed]
- Manteca, A.; Ye, J.; Sanchez, J.; Jensen, O.N. Phosphoproteome analysis of Streptomyces development reveals extensive protein phosphorylation accompanying bacterial differentiation. J. Proteome. Res. 2011, 10, 5481–5492. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Pinto, S.M.; Patil, A.H.; Advani, J.; Subba, P.; Kumar, M.; Sharma, J.; Dey, G.; Ravikumar, R.; Buggi, S.; et al. Quantitative proteomic and phosphoproteomic analysis of H37Ra and H37Rv strains of Mycobacterium tuberculosis. J. Proteome Res. 2017, 16, 1632–1645. [Google Scholar] [CrossRef]
- Albeldas, C.; Ganief, N.; Calder, B.; Nakedi, K.C.; Garnett, S.; Nel, A.J.M.; Blackburn, J.M.; Soares, N.C. Global proteome and phosphoproteome dynamics indicate novel mechanisms of vitamin C induced dormancy in Mycobacterium smegmatis. J. Proteom. 2018, 180, 1–10. [Google Scholar] [CrossRef]
- Liao, G.; Xie, L.; Li, X.; Cheng, Z.; Xie, J. Unexpected extensive lysine acetylation in the trump-card antibiotic producer Streptomyces roseosporus revealed by proteome-wide profiling. J. Proteom. 2014, 106, 260–269. [Google Scholar] [CrossRef]
- Huang, D.; Li, Z.H.; You, D.; Zhou, Y.; Ye, B.C. Lysine acetylproteome analysis suggests its roles in primary and secondary metabolism in Saccharopolyspora erythraea. Appl. Microbiol. Biotechnol. 2015, 99, 1399–1413. [Google Scholar] [CrossRef]
- Xie, L.; Wang, X.; Zeng, J.; Zhou, M.; Duan, X.; Li, Q.; Zhang, Z.; Luo, H.; Pang, L.; Li, W.; et al. Proteome-wide lysine acetylation profiling of the human pathogen Mycobacterium tuberculosis. Int. J. Biochem. Cell Biol. 2015, 59, 193–202. [Google Scholar] [CrossRef]
- Xu, J.Y.; Xu, Z.; Zhou, Y.; Ye, B.C. Lysine malonylome may affect the central metabolism and erythromycin biosynthesis pathway in Saccharopolyspora erythraea. J. Proteome Res. 2016, 15, 1685–1701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, P.; Ren, S.; Cheng, Z.; Zhao, G.; Zhao, W. ScCobB2-mediated lysine desuccinylation regulates protein biosynthesis and carbon metabolism in Streptomyces coelicolor. Mol. Cell Proteom. 2019, 18, 2003–2017. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xie, L.; Yang, Z.; Zhou, J.; Xie, J. Lysine succinylation of Mycobacterium tuberculosis isocitrate lyase (ICL) fine-tunes the microbial resistance to antibiotics. J. Biomol. Struct. Dyn. 2017, 35, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, Y.; Chen, Y.; Cheng, Z.; Gu, J.; Deng, J.; Bi, L.; Chen, C.; Mo, R.; Wang, X.; et al. Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis. Mol. Cell Proteom. 2015, 14, 796–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Wang, G.; Yu, Z.; Zhou, M.; Li, Q.; Huang, H.; Xie, J. Proteome-wide lysine glutarylation profiling of the Mycobacterium tuberculosis H37Rv. J. Proteome Res. 2016, 15, 1379–1385. [Google Scholar] [CrossRef]
- You, D.; Wang, M.M.; Ye, B.C. Acetyl-CoA synthetases of Saccharopolyspora erythraea are regulated by the nitrogen response regulator GlnR at both transcriptional and post-translational levels. Mol. Microbiol. 2017, 103, 845–859. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.Y.; Zhai, G.J.; Chen, C.; Bai, X.; Tian, S.S.; Hu, D.Q.; Fan, E.G.; Zhang, K. Protein lysine de-2-hydroxyisobutyrylation by CobB in prokaryotes. Sci. Adv. 2019, 5, eaaw6703. [Google Scholar] [CrossRef] [Green Version]
- Keenan, T.; Dowle, A.; Bates, R.; Smith, M. Characterization of the Streptomyces coelicolor glycoproteome reveals glycoproteins important for cell wall biogenesis. mBio 2019, 10, e01092. [Google Scholar] [CrossRef] [Green Version]
- Szirák, K.; Keserű, J.; Biró, S.; Schmelczer, I.; Barabás, G.; Penyige, A. Disruption of sco5461 gene coding for a mono-ADP-ribosyltransferase enzyme produces a conditional pleiotropic phenotype affecting morphological differentiation and antibiotic production in Streptomyces coelicolor. J. Microbiol. 2012, 50, 409–418. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef]
- Hong, S.Y.; Roze, L.V.; Linz, J.E. Oxidative stress-related transcription factors in the regulation of secondary metabolism. Toxins 2013, 5, 683–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.Y.; Xu, Y.; Xu, Z.; Zhai, L.H.; Ye, Y.; Zhao, Y.; Chu, X.; Tan, M.; Ye, B.C. Protein acylation is a general regulatory mechanism in biosynthetic pathway of acyl-CoA-derived natural products. Cell Chem. Biol. 2018, 25, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Macek, B.; Gnad, F.; Soufi, B.; Kumar, C.; Olsen, J.V.; Mijakovic, I.; Mann, M. Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell Proteom. 2008, 7, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, N.C.; Spät, P.; Krug, K.; Macek, B. Global dynamics of the Escherichia coli proteome and phosphoproteome during growth in minimal medium. J. Proteome Res. 2013, 12, 2611–2621. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashist, A.; Malhotra, V.; Sharma, G.; Tyagi, J.S.; Clark-Curtiss, J.E. Interplay of PhoP and DevR response regulators defines expression of the dormancy regulon in virulent Mycobacterium tuberculosis. J. Biol. Chem. 2018, 293, 16413–16425. [Google Scholar] [CrossRef] [Green Version]
- Henares, B.M.; Higgins, K.E.; Boon, E.M. Discovery of a nitric oxide responsive quorum sensing circuit in Vibrio harveyi. ACS Chem. Biol. 2012, 7, 1331–1336. [Google Scholar] [CrossRef]
- Martín, J.F.; Santos-Beneit, F.; Rodríguez-García, A.; Sola-Landa, A.; Smith, M.C.M.; Ellingsen, T.E.; Nieselt, K.; Burroughs, N.J.; Wellington, E.M.H. Transcriptomic studies of phosphate control of primary and secondary metabolism in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 2012, 95, 61–75. [Google Scholar] [CrossRef]
- McLean, T.C.; Lo, R.; Tschowri, N.; Hoskisson, P.A.; Al Bassam, M.M.; Hutchings, M.I.; Som, N.F. Sensing and responding to diverse extracellular signals: An updated analysis of the sensor kinases and response regulators of Streptomyces species. Microbiology 2019, 165, 929–952. [Google Scholar] [CrossRef]
- Ortet, P.; Whitworth, D.E.; Santaella, C.; Achouak, W.; Barakat, M. P2CS: Updates of the prokaryotic two-component systems database. Nucleic Acids Res. 2015, 43, D536–D541. [Google Scholar] [CrossRef] [Green Version]
- Capra, E.J.; Laub, M.T. Evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 2012, 66, 325–347. [Google Scholar] [CrossRef] [Green Version]
- Laub, M.T.; Goulian, M. Specificity in two-component signal transduction pathways. Annu. Rev. Genet. 2007, 41, 121–145. [Google Scholar] [CrossRef] [Green Version]
- Zschiedrich, C.P.; Keidel, V.; Szurmant, H. Molecular mechanisms of two-component signal transduction. J. Mol. Biol. 2016, 428, 3752–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, T.; Inouye, M. Ligand binding to the receptor domain regulates the ratio of kinase to phosphatase activities of the signaling domain of the hybrid Escherichia coli transmembrane receptor, Taz1. J. Mol. Biol. 1993, 232, 484–492. [Google Scholar] [CrossRef]
- Yang, Y.; Inouye, M. Requirement of both kinase and phosphatase activities of an Escherichia coli receptor (Taz1) for ligand-dependent signal transduction. J. Mol. Biol. 1993, 231, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Raivio, T.L.; Silhavy, T.J. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 1997, 179, 7724–7733. [Google Scholar] [CrossRef] [Green Version]
- Mascher, T.; Helmann, J.D.; Unden, G. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 910–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, W.J.; Luthra, A.; Zhu, G.; Radolf, J.D.; Malkowski, M.G.; Caimano, M.J. Structural characterization and modeling of the Borrelia burgdorferi hybrid histidine kinase Hk1 periplasmic sensor: A system for sensing small molecules associated with tick feeding. J. Struct. Biol. 2015, 192, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guła, G.; Dorotkiewicz-Jach, A.; Korzekwa, K.; Valvano, M.A.; Drulis-Kawa, Z. Complex signaling networks controlling dynamic molecular changes in Pseudomonas aeruginosa biofilm. Curr. Med. Chem. 2019, 26, 1979–1993. [Google Scholar] [CrossRef]
- Fiege, K.; Frankenberg-Dinkel, N. Thiol-based redox sensing in the methyltransferase associated sensor kinase RdmS in Methanosarcina acetivorans. Environ. Microbiol. 2019, 21, 1597–1610. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Y.; Chater, K.F.; Ou, H.Y.; Xu, H.H.; Deng, Z.; Tao, M. Large-scale transposition mutagenesis of Streptomyces coelicolor identifies hundreds of genes influencing antibiotic biosynthesis. Appl. Environ. Microbiol. 2017, 83, e02889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahrt, T.C.; Deretic, V. An essential two-component signal transduction system in Mycobacterium tuberculosis. J. Bacteriol. 2000, 182, 3832–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Som, N.F.; Heine, D.; Holmes, N.A.; Munnoch, J.T.; Chandra, G.; Seipke, R.F.; Hoskisson, P.A.; Wilkinson, B.; Hutchings, M.I. The conserved Actinobacterial two-component system MtrAB coordinates chloramphenicol production with sporulation in Streptomyces venezuelae NRRL B-65442. Front. Microbiol. 2017, 8, 1145. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhu, H.; Dang, F.; Zhang, W.; Qin, Z.; Yang, S.; Tan, H.; Lu, Y.; Jiang, W. Differential regulation of antibiotic biosynthesis by DraR-K, a novel two-component system in Streptomyces coelicolor. Mol. Microbiol. 2012, 85, 535–556. [Google Scholar] [CrossRef]
- Kwon, S.Y.; Kwon, H.J. The possible role of sco3388, a tmrB-like gene of Streptomyces coelicolor, in germination and stress survival of spores. J. Appl. Biol. Chem. 2013, 56, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Mast, Y.; Wang, J.; Zhang, W.W.; Zhao, G.P.; Wohlleben, W.; Lu, Y.H.; Jiang, W.H. Identification of two-component system AfsQ1/Q2 regulon and its cross-regulation with GlnR in Streptomyces coelicolor. Mol. Microbiol. 2013, 87, 30–48. [Google Scholar] [CrossRef]
- Allenby, N.E.; Laing, E.; Bucca, G.; Kierzek, A.M.; Smith, C.P. Diverse control of metabolism and other cellular processes in Streptomyces coelicolor by the PhoP transcription factor: Genome-wide identification of in vivo targets. Nucleic Acids Res. 2012, 40, 9543–9556. [Google Scholar] [CrossRef]
- Yepes, A.; Rico, S.; Rodríguez-García, A.; Santamaría, R.I.; Díaz, M. Novel two-component systems implied in antibiotic production in Streptomyces coelicolor. PLoS ONE 2011, 6, e19980. [Google Scholar] [CrossRef] [Green Version]
- Hesketh, A.; Hill, C.; Mokhtar, J.; Novotna, G.; Tran, N.; Bibb, M.; Hong, H.J. Genome-wide dynamics of a bacterial response to antibiotics that target the cell envelope. BMC Genom. 2011, 12, 226. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, H.; Horinouchi, S.; Kieser, H.M.; Hopwood, D.A.; Beppu, T. A putative two-component regulatory system involved in secondary metabolism in Streptomyces spp. J. Bacteriol. 1992, 174, 7585–7594. [Google Scholar] [CrossRef] [Green Version]
- Bishop, A.; Fielding, S.; Dyson, P.; Herron, P. Systematic insertional mutagenesis of a Streptomycete genome: A link between osmoadaptation and antibiotic production. Genome Res. 2004, 14, 893–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Jiang, W.; Lu, Y. A novel two-component system, GluR-GluK, involved in glutamate sensing and uptake in Streptomyces coelicolor. J. Bacteriol. 2017, 199, e00097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.M.; Chen, M.Y.; Shieh, Y.T.; Bibb, M.J.; Chen, C.W. The CutRS signal transduction system of Streptomyces lividans represses the biosynthesis of the polyketide antibiotic actinorhodin. Mol. Microbiol. 1996, 21, 1075–1085. [Google Scholar] [PubMed]
- Stancik, I.A.; Sestak, M.S.; Ji, B.; Axelson-Fisk, M.; Franjevic, D.; Jers, C.; Domazet-Loso, T.; Mijakovic, I. Serine/threonine protein kinases from bacteria, archaea and eukarya share a common evolutionary origin deeply rooted in the tree of life. J. Mol. Biol. 2018, 430, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef] [PubMed]
- Janczarek, M.; Vinardell, J.M.; Lipa, P.; Karas, M. Hanks-type serine/threonine protein kinases and phosphatases in bacteria: Roles in signaling and adaptation to various environments. Int. J. Mol. Sci. 2018, 19, 2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krupa, A.; Srinivasan, N. Diversity in domain architectures of ser/thr kinases and their homologues in prokaryotes. BMC Genom. 2005, 6, 129. [Google Scholar] [CrossRef] [Green Version]
- Grangeasse, C.; Cozzone, A.; Deutscher, J.; Mijakovic, I. Tyrosine phosphorylation: An emerging regulatory device of bacterial physiology. Trends Biochem. Sci. 2007, 32, 86–94. [Google Scholar] [CrossRef]
- Mijakovic, I.; Grangeasse, C.; Turgay, K. Exploring the diversity of protein modifications: Special bacterial phosphorylation systems. FEMS Microbiol. Rev. 2016, 40, 398–417. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.C.; Zheng, J.M.; She, Y.M.; Jia, Z.C. Structure of Escherichia coli tyrosine kinase Etk reveals a novel activation mechanism. EMBO J. 2008, 27, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Young, T.A.; Delagoutte, B.; Endrizzi, J.A.; Falick, A.M.; Alber, T. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for ser/thr protein kinases. Nat. Struct. Biol. 2003, 10, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Lombardía, M.; Pompeo, F.; Boitel, B.; Alzari, P.M. Crystal structure of the catalytic domain of the PknB serine/threonine kinase from Mycobacterium tuberculosis. J. Biol. Chem. 2003, 278, 13094–13100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Platig, J.; Cheng, T.Y.; Ahmed, S.; Skaf, Y.; Potluri, L.P.; Schwartz, D.; Steen, H.; Moody, D.B.; Husson, R.N. Protein kinases PknA and PknB independently and coordinately regulate essential Mycobacterium tuberculosis physiologies and antimicrobial susceptibility. PLoS Pathog. 2020, 16, e1008452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenstein, A.E.; MacGurn, J.A.; Baer, C.E.; Falick, A.M.; Cox, J.S.; Alber, T. M. tuberculosis ser/thr protein kinase D phosphorylates an anti-anti-sigma factor homolog. PLoS Pathog. 2007, 3, e49. [Google Scholar] [CrossRef]
- Mieczkowski, C.; Iavarone, A.T.; Alber, T. Auto-activation mechanism of the Mycobacterium tuberculosis PknB receptor ser/thr kinase. EMBO J. 2008, 27, 3186–3197. [Google Scholar] [CrossRef] [Green Version]
- Horinouchi, S. AfsR as an integrator of signals that are sensed by multiple serine/threonine kinases in Streptomyces coelicolor A3(2). J. Ind. Microbiol. Biotechnol. 2003, 30, 462–467. [Google Scholar] [CrossRef]
- Urabe, H.; Ogawara, H.; Motojima, K. Expression and characterization of Streptomyces coelicolor serine/threonine protein kinase PkaE. Biosci. Biotechnol. Biochem. 2015, 79, 855–862. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, Q.W.; Liu, Y.F.; Sun, S.F.; Chen, X.A.; Burchmore, R.; Burgess, K.; Li, Y.Q.; Mao, X.M. Multi-Layer Controls of Cas9 Activity Coupled With ATP Synthase Over-Expression for Efficient Genome Editing in Streptomyces. Front. Bioeng. Biotechnol. 2019, 7, 304. [Google Scholar] [CrossRef]
- Xu, J.Y.; You, D.; Leng, P.Q.; Ye, B.C. Allosteric regulation of a protein acetyltransferase in Micromonospora aurantiaca by the amino acids cysteine and arginine. J. Biol. Chem. 2014, 289, 27034–27045. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.X.; Liu, X.X.; Liu, W.B.; Ye, B.C. Identification and characterization of two types of amino acid-regulated acetyltransferases in Actinobacteria. Biosci. Rep. 2017, 37, BSR20170157. [Google Scholar] [CrossRef] [Green Version]
- McClure, J.J.; Inks, E.S.; Zhang, C.; Peterson, Y.K.; Li, J.; Chundru, K.; Lee, B.; Buchanan, A.; Miao, S.; Chou, C.J. Comparison of the deacylase and deacetylase activity of zinc-dependent HDACs. ACS Chem. Biol. 2017, 12, 1644–1655. [Google Scholar] [CrossRef]
- Wei, W.; Liu, X.; Chen, J.; Gao, S.; Lu, L.; Zhang, H.; Ding, G.; Wang, Z.; Chen, Z.; Shi, T.; et al. Class I histone deacetylases are major histone decrotonylases: Evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017, 27, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Colak, G.; Xie, Z.; Zhu, A.Y.; Dai, L.; Lu, Z.; Zhang, Y.; Wan, X.; Chen, Y.; Cha, Y.H.; Lin, H.; et al. Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell. Proteom. 2013, 12, 3509–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starai, V.J.; Celic, I.; Cole, R.N.; Boeke, J.D.; Escalante-Semerena, J.C. Sir2-dependent activation of acetyl-coa synthetase by deacetylation of active lysine. Science 2002, 298, 2390–2392. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Iesmantavicius, V.; Wagner, S.A.; Schölz, C.; Gummesson, B.; Beli, P.; Nyström, T.; Choudhary, C. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell. 2013, 51, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.M.; You, D.; Ye, B.C. Site-specific and kinetic characterization of enzymatic and nonenzymatic protein acetylation in bacteria. Sci. Rep. 2017, 7, 14790. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.I.; Lima, B.P.; Wolfe, A.J. Bacterial protein acetylation: The dawning of a new age. Mol. Microbiol. 2010, 77, 15–21. [Google Scholar] [CrossRef] [Green Version]
- AbouElfetouh, A.; Kuhn, M.L.; Hu, L.I.; Scholle, M.D.; Sorensen, D.J.; Sahu, A.K.; Becher, D.; Antelmann, H.; Mrksich, M.; Anderson, W.F.; et al. The E. coli sirtuin CobB shows no preference for enzymatic and nonenzymatic lysine acetylation substrate sites. Microbiologyopen 2015, 4, 66–83. [Google Scholar] [CrossRef] [Green Version]
- Erb, T.J.; Berg, I.A.; Brecht, V.; Muller, M.; Fuchs, G.; Alber, B.E. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 10631–10636. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; You, D.; Ye, B.C. Nitrogen regulator GlnR directly controls transcription of genes encoding lysine deacetylases in Actinobacteria. Microbiology 2017, 163, 1702–1710. [Google Scholar] [CrossRef]
- You, D.; Wang, M.M.; Yin, B.C.; Ye, B.C. Precursor supply for erythromycin biosynthesis: Engineering of propionate assimilation pathway based on propionylation modification. ACS Synth. Biol. 2019, 8, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.J.; Mintseris, J.; Ferreyra, J.; Gygi, S.P.; Darwin, K.H. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 2008, 322, 1104–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Niu, Y.; Liang, K.; Shen, G.; Cao, Q.; Yang, Y. Analysis of pupylation of Streptomyces hygroscopicus 5008 in vitro. Biochem. Biophys. Res. Commun. 2016, 474, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Akhter, Y.; Thakur, S. Targets of ubiquitin like system in mycobacteria and related actinobacterial species. Microbiol. Res. 2017, 204, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Satoh, Y.; Yanashima, K.; Matsui, T.; Dairi, T. Ergothioneine protects Streptomyces coelicolor A3(2) from oxidative stresses. J. Biosci. Bioeng. 2015, 120, 294–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boubakri, H.; Seghezzi, N.; Duchateau, M.; Gominet, M.; Kofronova, O.; Benada, O.; Mazodier, P.; Pernodet, J.L. The absence of pupylation (prokaryotic ubiquitin-like protein modification) affects morphological and physiological differentiation in Streptomyces coelicolor. J. Bacteriol. 2015, 197, 3388–3399. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wu, J.; Li, J.; Yang, H.; Tang, T.; Liang, H.; Zuo, M.; Wang, J.; Liu, H.; Liu, F.; et al. Host-mediated ubiquitination of a mycobacterial protein suppresses immunity. Nature 2020, 577, 682–688. [Google Scholar] [CrossRef]
- Salgame, P. Pupylation provides the punch as Mycobacterium tuberculosis battles the host macrophage. Cell Host Microbe 2008, 4, 415–416. [Google Scholar] [CrossRef] [Green Version]
- Bryk, R.; Gold, B.; Venugopal, A.; Singh, J.; Samy, R.; Pupek, K.; Cao, H.; Popescu, C.; Gurney, M.; Hotha, S.; et al. Selective killing of nonreplicating mycobacteria. Cell Host Microbe 2008, 3, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Compton, C.L.; Fernandopulle, M.S.; Nagari, R.T.; Sello, J.K. Genetic and proteomic analyses of pupylation in Streptomyces coelicolor. J. Bacteriol. 2015, 197, 2747–2753. [Google Scholar] [CrossRef] [Green Version]
- Worden, E.J.; Hoffmann, N.A.; Hicks, C.W.; Wolberger, C. Mechanism of cross-talk between H2B ubiquitination and H3 methylation by Dot1L. Cell 2019, 176, 1490–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, M.P.; Lee, M.J.; Ding, F.; Purbeck, C.; Kuhlman, B.; Dokholyan, N.V.; Dohlman, H.G. G protein mono-ubiquitination by the Rsp5 ubiquitin ligase. J. Biol. Chem. 2009, 284, 8940–8950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Q.; Wang, X.; Qiang, L.; Zhang, Y.; Ge, P.; Lu, Z.; Zhong, Y.; Li, B.; Wang, J.; Zhang, L.; et al. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat. Commun. 2019, 10, 1973. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Organism | Main Antibiotic | PTM | Functions | Reference |
|---|---|---|---|---|
| Streptomyces coelicolor | actinorhodin and clorobiocin | Phosphorylation | Modulating differentiation and secondary metabolism | [11] |
| Mycobacterium tuberculosis | none | Virulence, fatty acid biosynthesis and two-component regulatory system | [16] | |
| Mycobacterium smegmatis | none | Environmental adaptation, including dormancy and drug resistance | [17] | |
| Streptomyces roseosporus | daptomycin | Acetylation | Governing cellular processes, including secondary metabolites biosynthesis | [18] |
| Streptomyces coelicolor | actinorhodin and clorobiocin | Governing cellular processes, including secondary metabolites biosynthesis | [9] | |
| Saccharopolyspora erythraea | erythromycin | Central metabolism like protein synthesis, glycolysis, citric acid (TCA) cycle and a direct regulation in erythromycin synthesis | [19] | |
| Mycobacterium tuberculosis | none | Metabolism, persistence and virulence | [20] | |
| Saccharopolyspora erythraea | erythromycin | Malonylation | Central metabolism and erythromycin biosynthesis | [21] |
| Streptomyces coelicolor | actinorhodin and clorobiocin | Succinylation | Protein biosynthesis and carbon metabolism | [22] |
| Mycobacterium tuberculosis | none | Resistance to antibiotics | [23,24] | |
| Streptomyces roseosporus | daptomycin | Crotonylation | Governing cellular processes, including carbon catabolite repression and secondary metabolites biosynthesis | [14] |
| Mycobacterium tuberculosis | none | Glutarylation | Governing protein folding and metabolic process related with stress reaction | [25] |
| Streptomyces coelicolor | actinorhodin and clorobiocin | Pupylation | Protein degradation | [26] |
| Mycobacterium tuberculosis | none | Substance metabolism, toxic and antitoxic factors, cell wall and cell membrane components and pathogenicity | [27] | |
| Streptomyces coelicolor | actinorhodin and clorobiocin | O-glycosylation | Maintaining cell wall integrity and regulating enzyme function | [28] |
| Streptomyces coelicolor | actinorhodin and clorobiocin | ADP-ribosylation | Morphologic differentiation and antibiotic production | [29] |
| TCSs | Organism | Function | Reference |
|---|---|---|---|
| MacRS | S. coelicolor | Aerial mycelium formation/membrane integrity and/or other membrane-associated activities | [51] |
| MtrAB | M. tuberculosis | DNA replication and cell division | [52] |
| S. venezuelae | Antibiotic production, nutrient assimilation and aerial mycelium formation | [53] | |
| DraRK | S. coelicolor | Antibiotic production | [54] |
| TunRS | S. coelicolor | Cell wall metabolism and tmrB-like gene regulation | [55] |
| CssRS | S. lividans | Misfolded protein regulation | [56] |
| PhoPR | Streptomyces | Phosphate assimilation and secondary metabolism | [57] |
| AbrC1/2/3 | S. coelicolor | Antibiotic production | [58] |
| EsrSR | S. coelicolor | Cell envelope stress response | [59] |
| AfsQ1/2 | S. coelicolor | Antibiotic production | [60] |
| OsaABC | S. coelicolor | Osmotic stress response | [61] |
| GluRK | S. coelicolor | Glutamate sensor | [62] |
| CutRS | S. lividans | Actinorhodin biosynthesis repression | [63] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.-F.; Li, Y.-Q.; Mao, X.-M. Regulation of Protein Post-Translational Modifications on Metabolism of Actinomycetes. Biomolecules 2020, 10, 1122. https://doi.org/10.3390/biom10081122
Sun C-F, Li Y-Q, Mao X-M. Regulation of Protein Post-Translational Modifications on Metabolism of Actinomycetes. Biomolecules. 2020; 10(8):1122. https://doi.org/10.3390/biom10081122
Chicago/Turabian StyleSun, Chen-Fan, Yong-Quan Li, and Xu-Ming Mao. 2020. "Regulation of Protein Post-Translational Modifications on Metabolism of Actinomycetes" Biomolecules 10, no. 8: 1122. https://doi.org/10.3390/biom10081122
APA StyleSun, C. -F., Li, Y. -Q., & Mao, X. -M. (2020). Regulation of Protein Post-Translational Modifications on Metabolism of Actinomycetes. Biomolecules, 10(8), 1122. https://doi.org/10.3390/biom10081122