CB2 Receptors and Neuron–Glia Interactions Modulate Neurotoxicity Generated by MAGL Inhibition

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
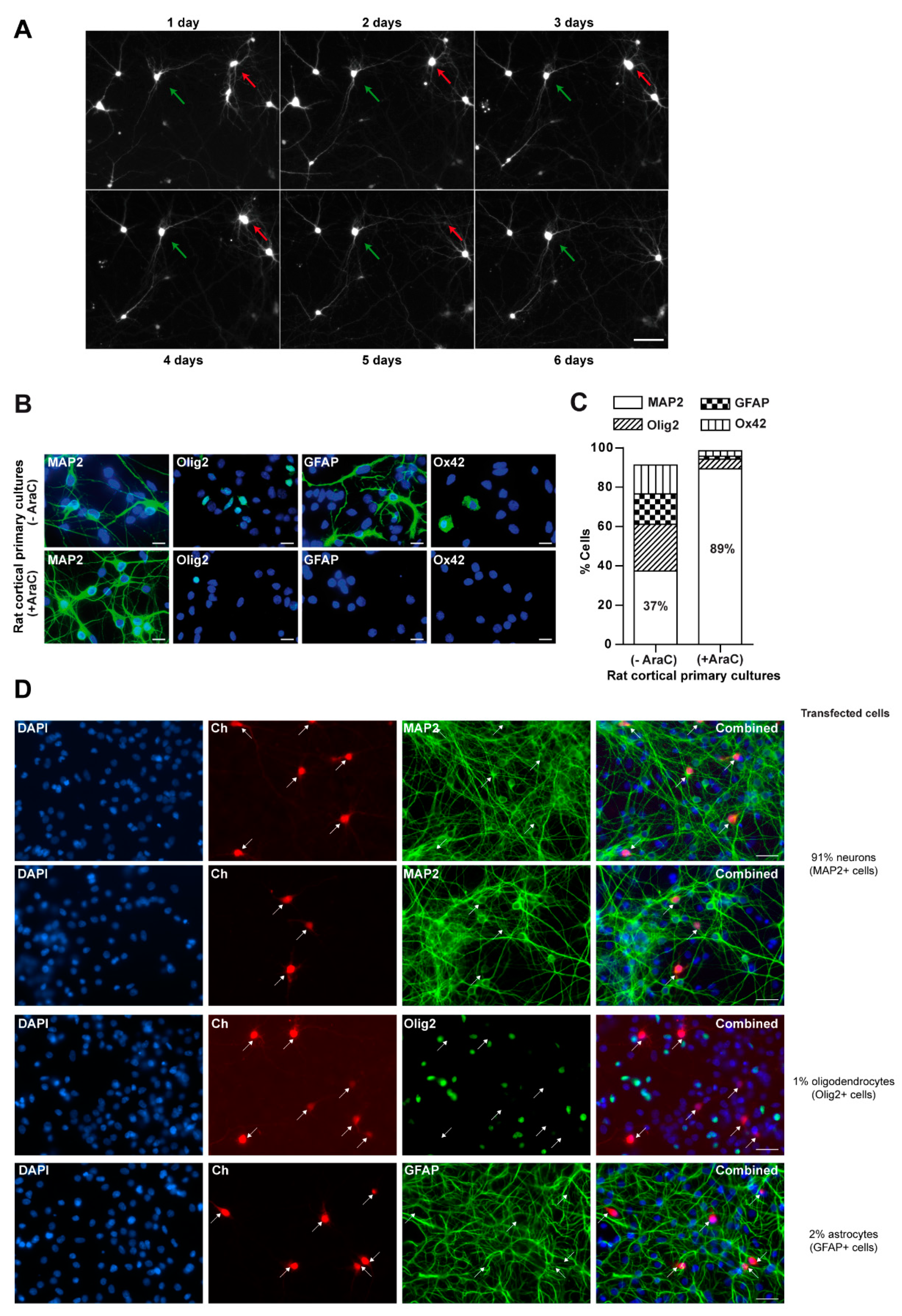
2.1. Rat Cortical Primary Cultures
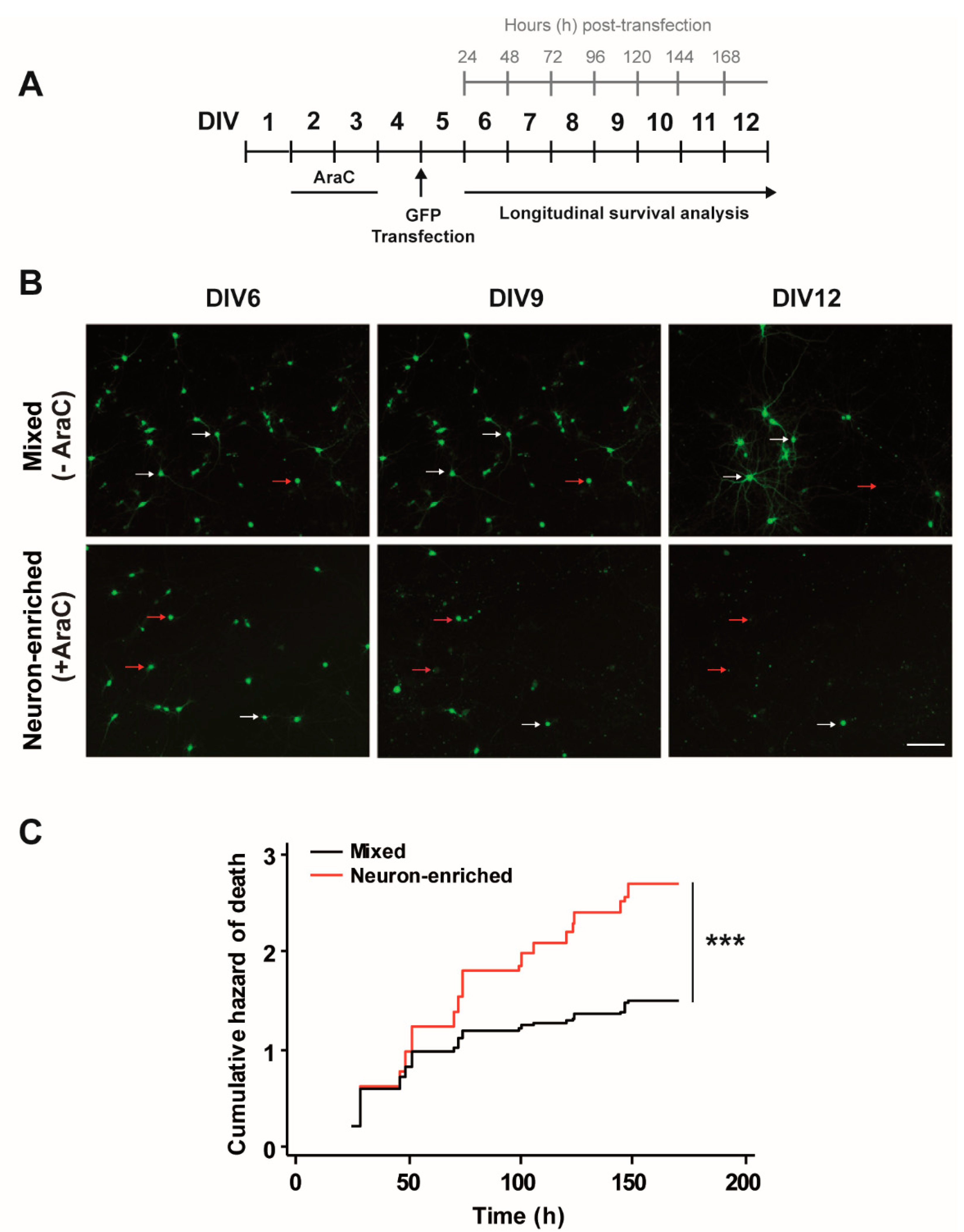
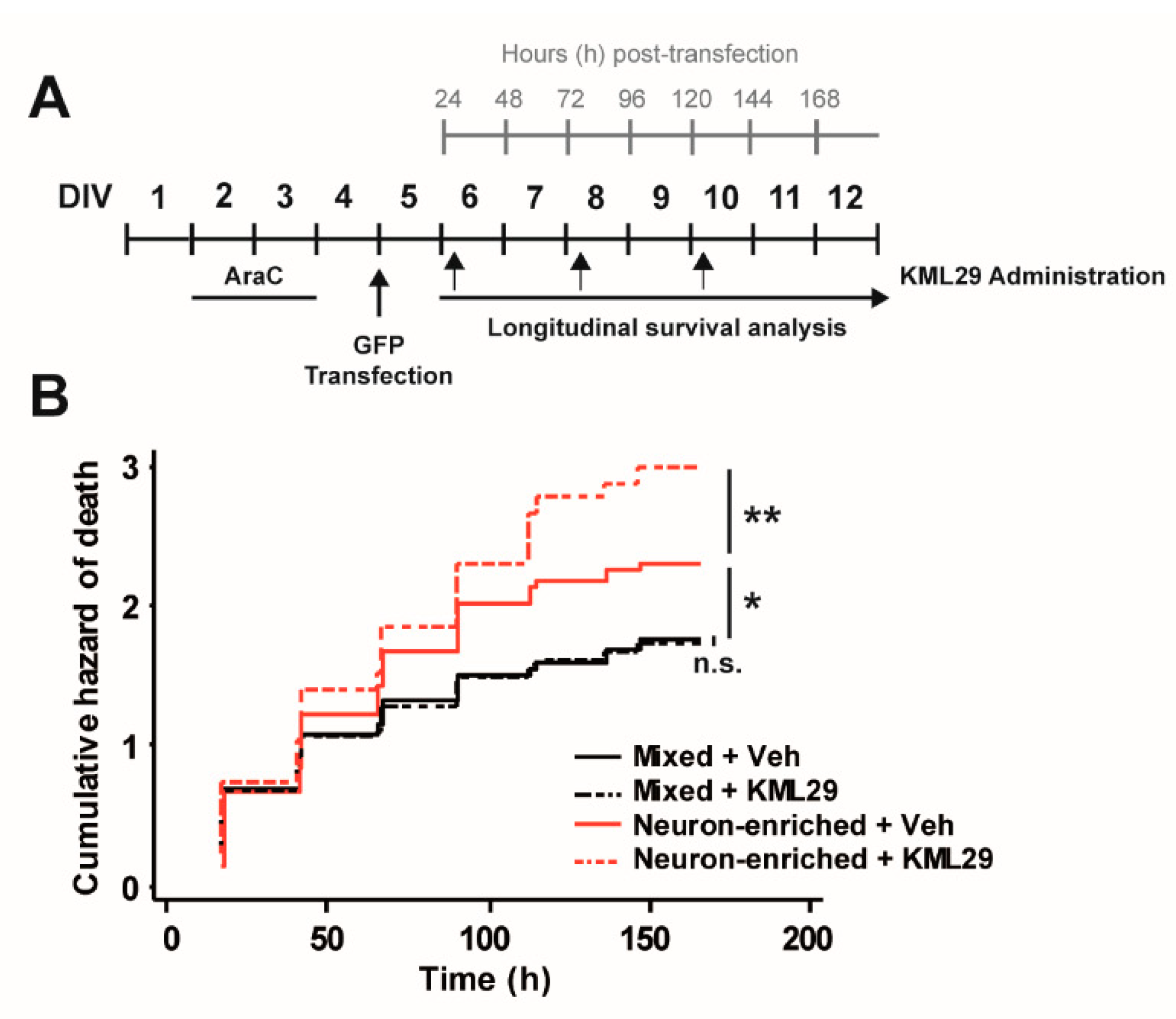
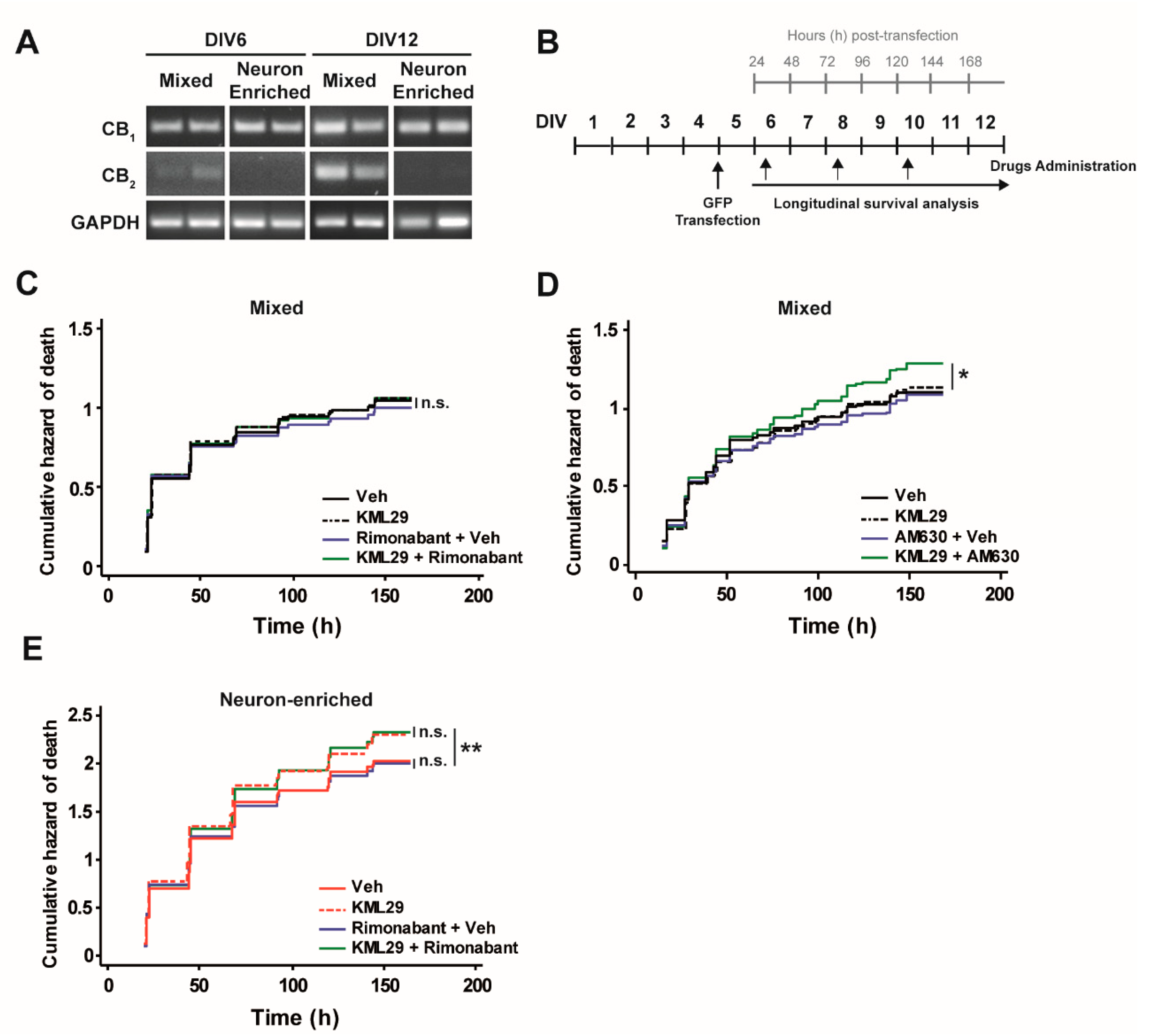
2.2. Lipofectamine Transfection of Rat Cortical Primary Cultures and Drug Treatments
2.3. Immunofluorescence
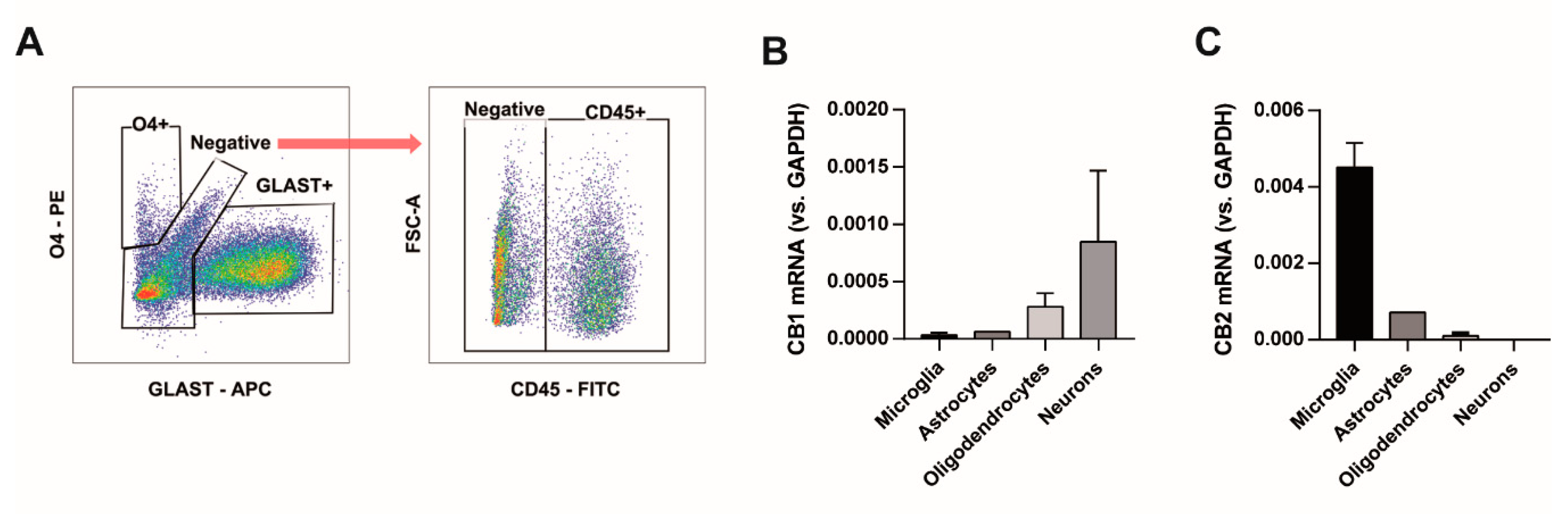
2.4. Fluorescence-Activated Cell Sorting
2.5. RNA Extraction and PCR Reaction
2.6. Automated Image Acquisition
2.7. Image Processing and Statistics
3. Results
3.1. Glial Cells Are Necessary for Neuronal Survival
3.2. Glial Cells Counteract the Neurotoxic Effect of MAGL Inhibition through CB2 Receptors.
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoylglycerol |
| AraC | Cytosine β-D-Arabinofuranoside hydrochloride |
| CB1 receptor | Cannabinoid receptor type 1 |
| CB2 receptor | Cannabinoid receptor type 2 |
| CD45 | Cluster of differentiation molecule 45 |
| GFP | Green fluorescent protein |
| GLAST | Glutamate-aspartate transporter |
| MAGL | Monoacylglycerol lipase |
| MPP+ | 1-methyl-4-phenylpyridinium |
References
- Martín, R.; Bajo-Grañeras, R.; Moratalla, R.; Perea, G.; Araque, A. Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 2015, 349, 730–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarrete, M.; Araque, A. Endocannabinoids Mediate Neuron-Astrocyte Communication. Neuron 2008, 57, 883–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarrete, M.; Araque, A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 2010, 68, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.-Y.; Strassle, B.W.; Lu, P.; et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef]
- Viader, A.; Ogasawara, D.; Joslyn, C.M.; Sanchez-Alavez, M.; Mori, S.; Nguyen, W.; Conti, B.; Cravatt, B.F. A chemical proteomic atlas of brain serine hydrolases identifies cell type-specific pathways regulating neuroinflammation. Elife 2016, 18, e12345. [Google Scholar] [CrossRef]
- Baggelaar, M.P.; Maccarrone, M.; van der Stelt, M. 2-Arachidonoylglycerol: A signaling lipid with manifold actions in the brain. Prog. Lipid Res. 2018, 71, 1–17. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Savinainen, J.R.; Järvinen, T.; Laine, K.; Laitinen, J.T. Despite sabstantial degradation, 2-arachidonoylglycerol is a potent full efficacy agonist mediating CB1 receptor-dependent g-protein activation in rat cerebellar membranes. Br. J. Pharmacol. 2001, 134, 664–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, T.; Kondo, S.; Kishimoto, S.; Miyashita, T.; Nakane, S.; Kodaka, T.; Suhara, Y.; Takayama, H.; Waku, K. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J. Biol. Chem. 2000, 275, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; De Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar] [PubMed]
- Navarrete, F.; García-Gutiérrez, M.S.; Aracil-Fernández, A.; Lanciego, J.L.; Manzanares, J. Cannabinoid CB1 and CB2 Receptors, and Monoacylglycerol Lipase Gene Expression Alterations in the Basal Ganglia of Patients with Parkinson’s Disease. Neurotherapeutics 2018, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viscomi, M.T.; Oddi, S.; Latini, L.; Pasquariello, N.; Florenzano, F.; Bernardi, G.; Molinari, M.; Maccarrone, M. Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J. Neurosci. 2009, 29, 4564–4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Núñez, E.; Benito, C.; Pazos, M.R.; Barbachano, A.; Fajardo, O.; González, S.; Tolón, R.M.; Romero, J. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: An Immunohistochemical Study. Synapse 2004, 53, 208–213. [Google Scholar] [CrossRef]
- Ramírez, B.G.; Blázquez, C.; Gómez Del Pulgar, T.; Guzmán, M.; De Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Cabral, G.A.; Griffin-Thomas, L.T. Emerging role of the cannabinoid receptor CB 2 in immune regulation: Therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 2009, 11, e3. [Google Scholar] [CrossRef] [Green Version]
- Benito, C.; Tolón, R.M.; Pazos, M.R.; Núñez, E.; Castillo, A.I.; Romero, J. Cannabinoid CB 2 receptors in human brain inflammation. Br. J. Pharmacol. 2008, 153, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [Green Version]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [Green Version]
- Long, J.Z.; Nomura, D.K.; Cravatt, B.F. Characterization of Monoacylglycerol Lipase Inhibition Reveals Differences in Central and Peripheral Endocannabinoid Metabolism. Chem. Biol. 2009, 16, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taschler, U.; Radner, F.P.W.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertwee, R.G. Targeting the endocannabinoid system with cannabinoid receptor agonists: Pharmacological strategies and therapeutic possibilities. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3353–3363. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, S.G.; Long, J.Z.; O’Neal, S.T.; Abdullah, R.A.; Poklis, J.L.; Boger, D.L.; Cravatt, B.F.; Lichtman, A.H. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J. Pharmacol. Exp. Ther. 2009, 330, 902–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, J.; Guijarro, A.; Piomelli, D.; Hohmann, A.G. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br. J. Pharmacol. 2011, 163, 1464–1478. [Google Scholar] [CrossRef] [Green Version]
- Woodhams, S.G.; Wong, A.; Barrett, D.A.; Bennett, A.J.; Chapman, V.; Alexander, S.P.H. Spinal administration of the monoacylglycerol lipase inhibitor JZL184 produces robust inhibitory effects on nociceptive processing and the development of central sensitization in the rat. Br. J. Pharmacol. 2012, 167, 1609–1619. [Google Scholar] [CrossRef]
- Ghosh, S.; Wise, L.E.; Chen, Y.; Gujjar, R.; Mahadevan, A.; Cravatt, B.F.; Lichtman, A.H. The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci. 2013, 92, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Griebel, G.; Pichat, P.; Beeske, S.; Leroy, T.; Redon, N.; Jacquet, A.; Françon, D.; Bert, L.; Even, L.; Lopez-Grancha, M.; et al. Selective blockade of the hydrolysis of the endocannabinoid 2-arachidonoylglycerol impairs learning and memory performance while producing antinociceptive activity in rodents. Sci. Rep. 2015, 5, 7642. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Puighermanal, E.; Pastor, A.; De La Torre, R.; Maldonado, R.; Ozaita, A. Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol. Psychiatry 2011, 70, 479–486. [Google Scholar] [CrossRef]
- Zhong, P.; Wang, W.; Pan, B.; Liu, X.; Zhang, Z.; Long, J.Z.; Zhang, H.T.; Cravatt, B.F.; Liu, Q.S. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology 2014, 39, 1763–1776. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.J.; Stuhr, K.L.; Hutz, M.J.; Raff, H.; Hillard, C.J. Endocannabinoid signaling in hypothalamic-pituitary-adrenocortical axis recovery following stress: Effects of indirect agonists and comparison of male and female mice. Pharmacol. Biochem. Behav. 2014, 117, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflammation 2015, 12. [Google Scholar] [CrossRef] [Green Version]
- Katz, P.S.; Sulzer, J.K.; Impastato, R.A.; Teng, S.X.; Rogers, E.K.; Molina, P.E. Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J. Neurotrauma 2015, 32, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Suárez, D.; Celorrio, M.; Riezu-Boj, J.I.; Ugarte, A.; Pacheco, R.; González, H.; Oyarzabal, J.; Hillard, C.J.; Franco, R.; Aymerich, M.S. The monoacylglycerol lipase inhibitor JZL184 is neuroprotective and alters glial cell phenotype in the chronic MPTP mouse model. Neurobiol. Aging 2014, 35, 2603–2616. [Google Scholar] [CrossRef]
- Martínez-Torres, S.; Cutando, L.; Pastor, A.; Kato, A.; Sakimura, K.; de la Torre, R.; Valjent, E.; Maldonado, R.; Kano, M.; Ozaita, A. Monoacylglycerol lipase blockade impairs fine motor coordination and triggers cerebellar neuroinflammation through cyclooxygenase-2. Brain. Behav. Immun. 2019, 81, 399–409. [Google Scholar] [CrossRef]
- Aymerich, M.S.; Rojo-Bustamante, E.; Molina, C.; Celorrio, M.; Sánchez-Arias, J.A.; Franco, R. Neuroprotective Effect of JZL184 in MPP+-Treated SH-SY5Y Cells Through CB2 Receptors. Mol. Neurobiol. 2016, 53, 2312–2319. [Google Scholar] [CrossRef]
- Arrasate, M.; Finkbeiner, S. Automated microscope system for determining factors that predict neuronal fate. Proc. Natl. Acad. Sci. USA. 2005, 102, 3840–3845. [Google Scholar] [CrossRef] [Green Version]
- Íñigo-Marco, I.; Valencia, M.; Larrea, L.; Bugallo, R.; Martínez-Goikoetxea, M.; Zuriguel, I.; Arrasate, M. E46K α-synuclein pathological mutation causes cell-autonomous toxicity without altering protein turnover or aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, E8274–E8283. [Google Scholar] [CrossRef] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinueza-Gavilanes, R.; Íñigo-Marco, I.; Larrea, L.; Lasa, M.; Carte, B.; Santamaría, E.; Fernández-Irigoyen, J.; Bugallo, R.; Aragón, T.; Aldabe, R.; et al. N-terminal acetylation mutants affect alpha-synuclein stability, protein levels and neuronal toxicity. Neurobiol. Dis. 2020, 137. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Niphakis, M.J.; Lum, K.M.; Cognetta, A.B.; Wang, C.; Matthews, M.L.; Niessen, S.; Buczynski, M.W.; Parsons, L.H.; Cravatt, B.F. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem. Biol. 2012, 19, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem 2005, 95, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 2014, 62, 1377–1391. [Google Scholar] [CrossRef]
- Verdonk, F.; Roux, P.; Flamant, P.; Fiette, L.; Bozza, F.A.; Simard, S.; Lemaire, M.; Plaud, B.; Shorte, S.L.; Sharshar, T.; et al. Phenotypic clustering: A novel method for microglial morphology analysis. J. Neuroinflammation 2016, 13, 153. [Google Scholar] [CrossRef]
- Bisogno, T.; Oddi, S.; Piccoli, A.; Fazio, D.; Maccarrone, M. Type-2 cannabinoid receptors in neurodegeneration. Pharmacol. Res. 2016, 111, 721–730. [Google Scholar] [CrossRef]
- Witting, A.; Möller, T. Microglia cell culture: A primer for the novice. Methods Mol. Biol. 2011, 758, 49–66. [Google Scholar] [CrossRef]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflammation 2012, 9. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef]
- Tang, J.; Miao, H.; Jiang, B.; Chen, Q.; Tan, L.; Tao, Y.; Zhang, J.; Gao, F.; Feng, H.; Zhu, G.; et al. A selective CB2R agonist (JWH133) restores neuronal circuit after Germinal Matrix Hemorrhage in the preterm via CX3CR1+ microglia. Neuropharmacology 2017, 119, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Hernangómez, M.; Mestre, L.; Correa, F.G.; Loría, F.; Mecha, M.; Iñigo, P.M.; Docagne, F.; Williams, R.O.; Borrell, J.; Guaza, C. CD200-CD200R1 interaction contributes to neuroprotective effects of anandamide on experimentally induced inflammation. Glia 2012, 60, 1437–1450. [Google Scholar] [CrossRef] [Green Version]
- Aymerich, M.S.; Aso, E.; Abellanas, M.A.; Tolon, R.M.; Ramos, J.A.; Ferrer, I.; Romero, J.; Fernández-Ruiz, J. Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018, 157, 67–84. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Provider (Catalog Number) | Type | Dilution |
|---|---|---|---|
| MAP2 | Sigma (M1406) Clon AP20 | Primary mouse monoclonal | 1:1000 |
| GFAP | Sigma (G3893) | Primary mouse monoclonal | 1:1000 |
| Olig2 | Merck-Millipore (MABN50, clon 211F1.1) | Primary mouse monoclonal | 1:200 |
| Ox-42 | BD Pharmingen (550299) | Primary mouse monoclonal | 1:50 |
| Cy5-conjugated goat anti mouse IgG | Jackson Immunoresearch (115-175-166) | Secondary goat Cy5-conjugated | 1:500 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojo-Bustamante, E.; Íñigo-Marco, I.; Abellanas, M.A.; Vinueza-Gavilanes, R.; Baltanás, A.; Luquin, E.; Arrasate, M.; Aymerich, M.S. CB2 Receptors and Neuron–Glia Interactions Modulate Neurotoxicity Generated by MAGL Inhibition. Biomolecules 2020, 10, 1198. https://doi.org/10.3390/biom10081198
Rojo-Bustamante E, Íñigo-Marco I, Abellanas MA, Vinueza-Gavilanes R, Baltanás A, Luquin E, Arrasate M, Aymerich MS. CB2 Receptors and Neuron–Glia Interactions Modulate Neurotoxicity Generated by MAGL Inhibition. Biomolecules. 2020; 10(8):1198. https://doi.org/10.3390/biom10081198
Chicago/Turabian StyleRojo-Bustamante, Estefania, Ignacio Íñigo-Marco, Miguel Angel Abellanas, Rodrigo Vinueza-Gavilanes, Ana Baltanás, Esther Luquin, Montserrat Arrasate, and Maria S. Aymerich. 2020. "CB2 Receptors and Neuron–Glia Interactions Modulate Neurotoxicity Generated by MAGL Inhibition" Biomolecules 10, no. 8: 1198. https://doi.org/10.3390/biom10081198
APA StyleRojo-Bustamante, E., Íñigo-Marco, I., Abellanas, M. A., Vinueza-Gavilanes, R., Baltanás, A., Luquin, E., Arrasate, M., & Aymerich, M. S. (2020). CB2 Receptors and Neuron–Glia Interactions Modulate Neurotoxicity Generated by MAGL Inhibition. Biomolecules, 10(8), 1198. https://doi.org/10.3390/biom10081198