Immune Monitoring upon Treatment with Biologics in Sjögren’s Syndrome: The What, Where, When, and How


Abstract
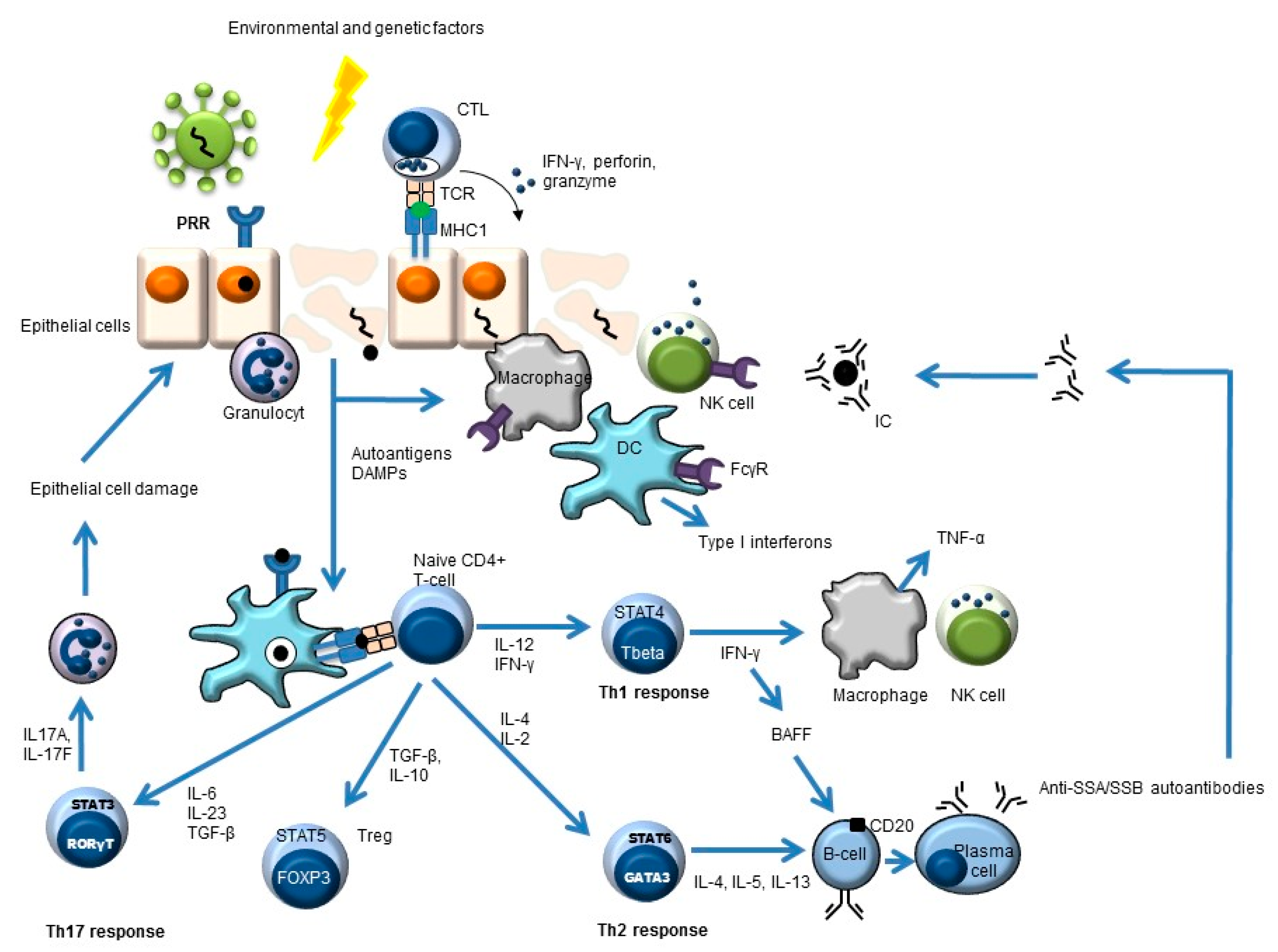
:1. Primary Sjögren’s Syndrome
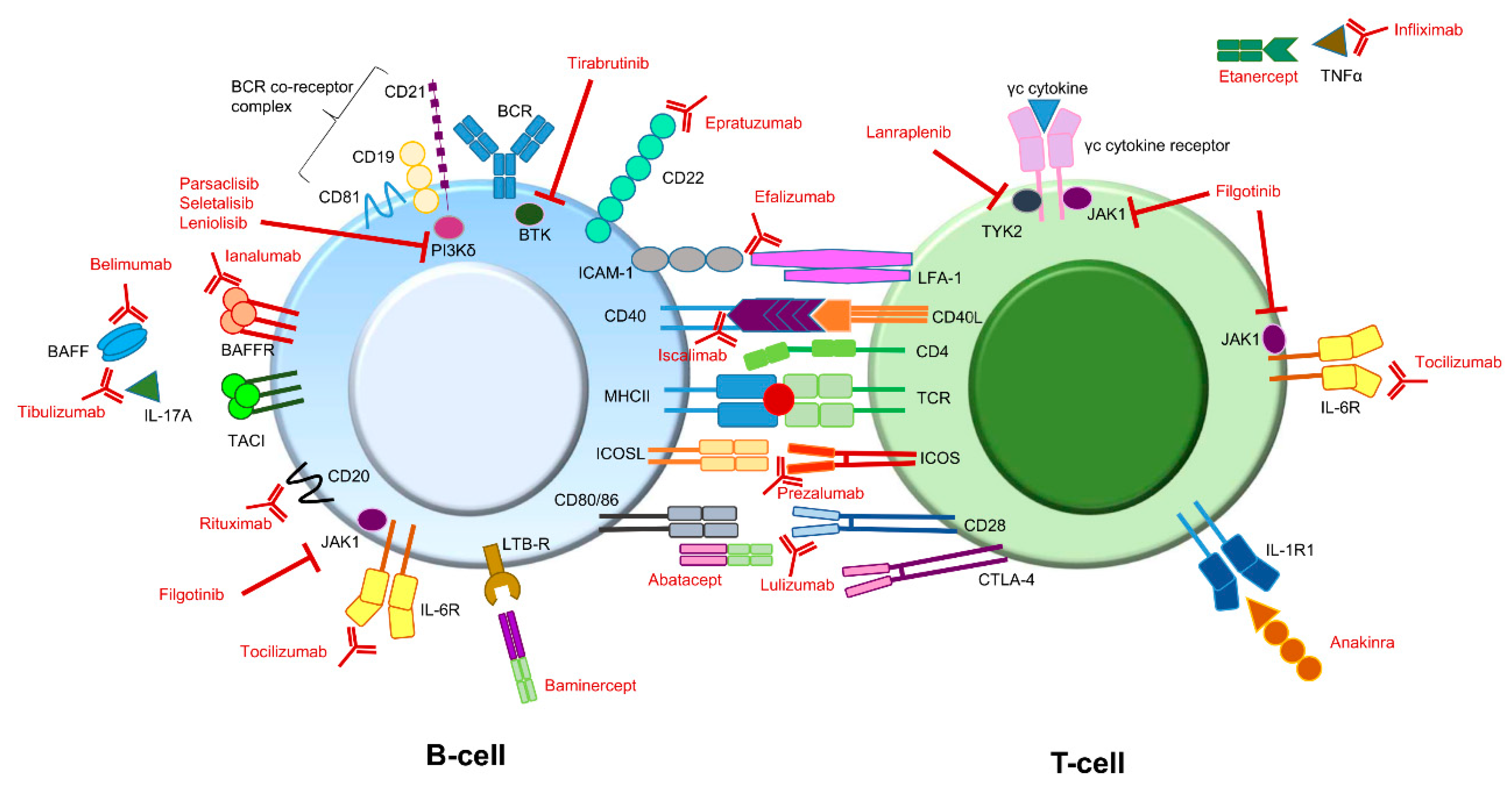
2. Targets of Biologics in pSS—The “What”
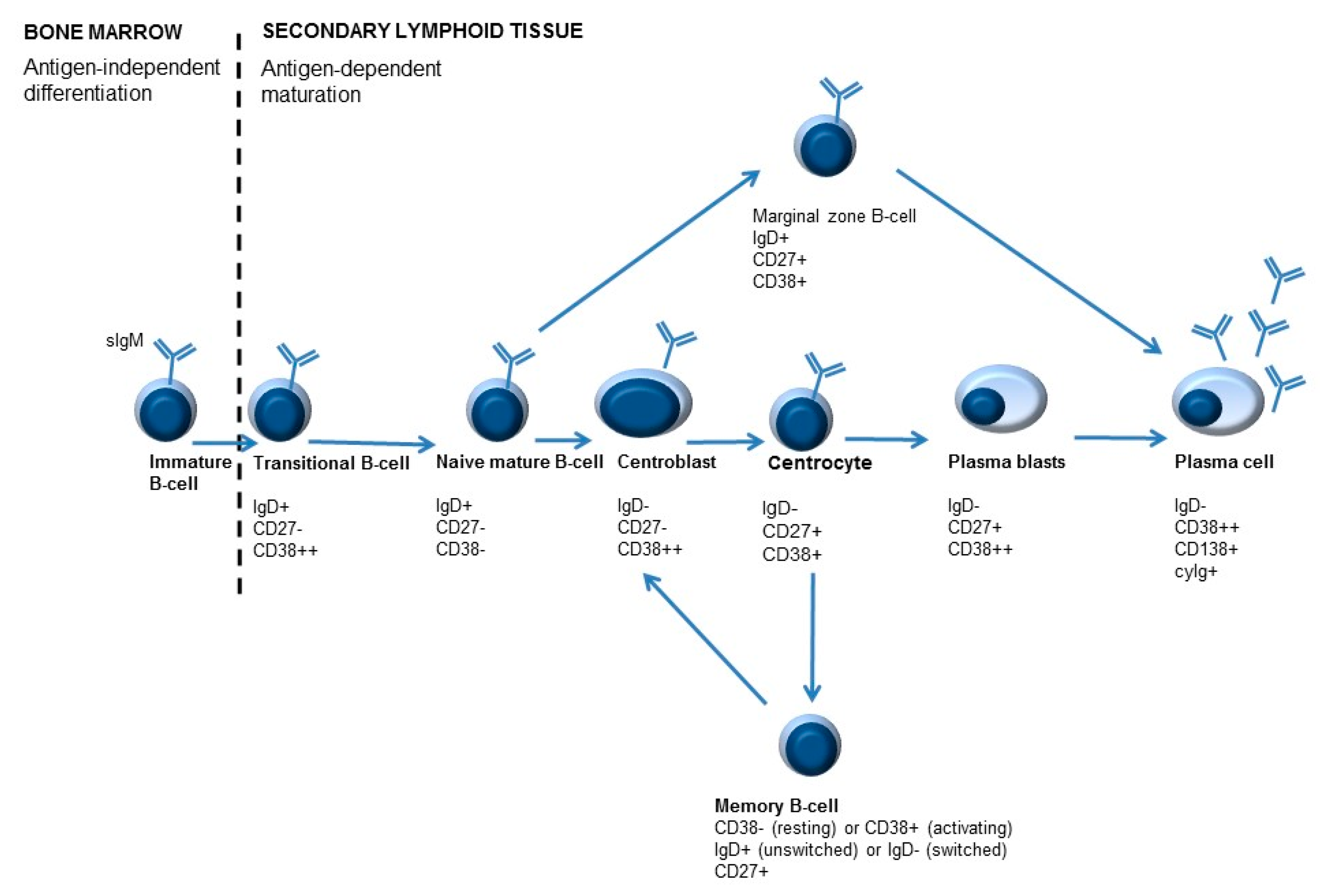
3. Immune Monitoring—The “Where”
4. Immune Monitoring—The “When”
5. Immune Monitoring—The “How”
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Both, T.; Dalm, V.A.H.; van Hagen, P.M.; van Daele, P. Reviewing primary Sjögren’s syndrome: Beyond the dryness—From pathophysiology to diagnosis and treatment. Int. J. Med. Sci. 2017, 14, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League against Rheumatism classification criteria for primary Sjögren’s syndrome: A consensus and data-driven methodology involving three international patient cohorts. Ann. Rheum. Dis. 2017, 76, 9–16. [Google Scholar] [CrossRef] [PubMed]
- De Wolff, L.; Arends, S.; van Nimwegen, J.F.; Bootsma, H. Ten years of the ESSDAI: Is it fit for purpose? Clin. Exp. Rheumatol. 2020, 126, 283–290. [Google Scholar]
- Seror, R.; Bowman, S.J.; Brito-Zeron, P.; Theander, E.; Bootsma, H.; Tzioufas, A.; Gottenberg, J.-E.; Ramos-Casals, M.; Dörner, T.; Ravaud, P.; et al. EULAR Sjögren’s syndrome disease activity index (ESSDAI): A user guide. RMD Open 2015, 1, e000022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seror, R.; Theander, E.; Brun, J.G.; Ramos-Casals, M.; Valim, V.; Dörner, T.; Bootsma, H.; Tzioufas, A.; Solans-Laqué, R.; Mandl, T.; et al. Validation of EULAR primary Sjögren’s syndrome disease activity (ESSDAI) and patient indexes (ESSPRI). Ann. Rheum. Dis. 2015, 74, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Seror, R.; Ravaud, P.; Mariette, X.; Bootsma, H.; Theander, E.; Hansen, A.; Ramos-Casals, M.; Doerner, T.; Bombardieri, S.; Hachulla, E.; et al. EULAR Sjögren’s syndrome patient reported index (ESSPRI): Development of a consensus patient index for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2011, 70, 968–972. [Google Scholar] [CrossRef]
- Cornec, D.; Devauchelle-Pensec, V.; Tobón, G.J.; Pers, J.-O.; Jousse-Joulin, S.; Saraux, A. B cells in Sjögren’s syndrome: From pathophysiology to diagnosis and treatment. J. Autoimmun. 2012, 39, 161–167. [Google Scholar] [CrossRef]
- Tannenbaum, H.; Pinkus, G.S.; Anderson, L.G.; Schur, P.H. Immunologic characterization of the mononuclear cell infiltrates in rheumatoid synovia, in rheumatoid nodules, and in lip biopsies from patients with Sjögren’s syndrome. Arthritis Rheum. 1975, 18, 305–314. [Google Scholar] [CrossRef]
- Henkin, R.I. Primary Sjögren’s syndrome. N. Engl. J. Med. 2018, 379, 96–97. [Google Scholar]
- Gandolfo, S.; De Vita, S. Emerging drugs for primary Sjögren’s syndrome. Expert Opin. Emerg. Drugs 2019, 24, 121–132. [Google Scholar] [CrossRef]
- Slobbe, R.L.; Pruijn, G.J.M.; Damen, W.G.M.; van der Kemp, J.W.C.M.; van Venrooij, W.J. Detection and occurrence of the 60- and 52-kD Ro (SS-A) antigens and of autoantibodies against these proteins. Clin. Exp. Immunol. 1991, 86, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Fasano, S.; Mauro, D.; Macaluso, F.; Xiao, F.; Zhao, Y.; Lu, L.; Guggino, G.; Ciccia, F. Pathogenesis of primary Sjogren’s syndrome beyond B lymphocytes. Clin. Exp. Rheumatol. 2020, 126, 315–323. [Google Scholar]
- Blokland, S.L.M.; van Vliet-Moret, F.M.; Hillen, M.R.; Pandit, A.; Goldschmeding, R.; Kruize, A.A.; Bouma, G.; van Maurik, A.; Olek, S.; Hoffmueller, U.; et al. Epigenetically quantified immune cells in salivary glands of Sjögren’s syndrome patients: A novel tool that detects robust correlations of T follicular helper cells with immunopathology. Rheumatology 2020, 59, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ming, B.; Zhang, C.; Deng, X.; Li, P.; Wei, Z.; Xia, Y.; Jiang, K.; Ye, H.; Ma, W.; et al. IL-2 inhibition of Th17 generation rather than induction of treg cells is impaired in primary Sjögren’s syndrome patients. Front. Immunol. 2018, 9, 1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hänninen, A.; Nordström, D.C. Activation of plasmacytoid dendritic cells by apoptotic particles—Mechanism for the loss of immunological tolerance in Sjögren’s syndrome. Clin. Exp. Immunol. 2017, 191, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Ushio, A.; Arakaki, R.; Otsuka, K.; Yamada, A.; Tsunematsu, T.; Kudo, Y.; Aota, K.; Azuma, M.; Ishimaru, N. CCL22-producing resident macrophages enhance T cell response in Sjögren’s syndrome. Front. Immunol. 2018, 9, 2594. [Google Scholar] [CrossRef]
- Vivino, F.B.; Carsons, S.E.; Foulks, G.; Daniels, T.E.; Parke, A.; Brennan, M.T.; Forstot, S.L.; Scofield, R.H.; Hammitt, K.M. New treatment guidelines for Sjögren’s disease. Rheum. Dis. Clin. N. Am. 2016, 42, 531–551. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Casals, M.; Brito-Zerón, P.; Bombardieri, S.; Bootsma, H.; De Vita, S.; Dörner, T.; Fisher, B.A.; Gottenberg, J.-E.; Hernández-Molina, G.; Kocher, A.; et al. EULAR recommendations for the management of Sjögren’s syndrome with topical and systemic therapies. Ann. Rheum. Dis. 2020, 79, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Manfre, V.; Cafaro, G.; Riccucci, I.; Zabotti, A.; Perricone, C.; Bootsma, H.; De Vita, S.; Bartoloni, E. One year in review 2020: Comorbidities, diagnosis and treatment of primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2020, 126, 10–22. [Google Scholar]
- Felten, R.; Scher, F.; Sibilia, J.; Gottenberg, J.-E.; Arnaud, L. The pipeline of targeted therapies under clinical development for primary Sjögren’s syndrome: A systematic review of trials. Autoimmun. Rev. 2019, 18, 576–582. [Google Scholar] [CrossRef]
- Sacco, K.A.; Abraham, R.S. Consequences of B-cell-depleting therapy: Hypogammaglobulinemia and impaired B-cell reconstitution. Immunotherapy 2018, 10, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, present, and future of rituximab—The world’s first oncology monoclonal antibody therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.-M.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.-E.; Chiche, L.; et al. Treatment of primary Sjögren syndrome with rituximab. Ann. Intern. Med. 2014, 160, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Bowman, S.J.; Everett, C.C.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.-F.; Pease, C.T.; Price, E.J.; Sutcliffe, N.; Gendi, N.S.T.; et al. Randomized controlled trial of rituximab and cost-effectiveness analysis in treating fatigue and oral dryness in primary Sjögren’s syndrome. Arthritis Rheumatol. 2017, 69, 1440–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, S.; Isenberg, D.A. Present and novel biologic drugs in primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2019, 118, 167–174. [Google Scholar]
- Álvarez-Rivas, N.; Sang-Park, H.; Del Campo, P.D.; Fernández-Castro, M.; Corominas, H.; Andreu, J.L.; Navarro-Compán, V. Efficacy of belimumab in primary Sjögren’s syndrome: A systematic review. Reumatol. Clin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Seror, R.; Quartuccio, L.; Baron, G.; Salvin, S.; Fabris, M.; Desmoulins, F.; Nocturne, G.; Ravaud, P.; De Vita, S. Efficacy and safety of belimumab in primary Sjögren’s syndrome: Results of the BELISS open-label phase II study. Ann. Rheum. Dis. 2015, 74, 526–531. [Google Scholar] [CrossRef]
- Castigli, E.; Wilson, S.A.; Scott, S.; Dedeoglu, F.; Xu, S.; Lam, K.-P.; Bram, R.J.; Jabara, H.; Geha, R. TACI and BAFF-R mediate isotype switching in B cells. J. Exp. Med. 2005, 201, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Dörner, T.; Posch, M.G.; Li, Y.; Petricoul, O.; Cabanski, M.; Milojevic, J.M.; Kamphausen, E.; Valentin, M.-A.; Simonett, C.; Mooney, L.; et al. Treatment of primary Sjögren’s syndrome with ianalumab (VAY736) targeting B cells by BAFF receptor blockade coupled with enhanced, antibody-dependent cellular cytotoxicity. Ann. Rheum. Dis. 2019, 78, 641–647. [Google Scholar] [CrossRef]
- Steinfeld, S.D.; Tant, L.; Burmester, G.-R.; Teoh, N.; Wegener, W.A.; Goldenberg, D.M.; Pradier, O. Epratuzumab (humanised anti-CD22 antibody) in primary Sjögren’s syndrome: An open-label phase I/II study. Arthritis Res. Ther. 2006, 8, R129. [Google Scholar] [CrossRef] [Green Version]
- St. Clair, E.W.; Baer, A.N.; Wei, C.; Noaiseh, G.; Parke, A.; Coca, A.; Utset, T.O.; Genovese, M.C.; Wallace, D.J.; McNamara, J.; et al. Clinical efficacy and safety of baminercept, a lymphotoxin β receptor fusion protein, in primary Sjögren’s syndrome. Arthritis Rheumatol. 2018, 70, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vignali, D.A.A. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.A.; Moutsopoulos, H.M. Sjögren’s syndrome: Old and new therapeutic targets. J. Autoimmun. 2020, 110, 102364. [Google Scholar]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 costimulation: From mechanism to therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontarini, E.; Verstappen, G.M.; Grigoriadou, S.; Kroese, F.G.M.; Bootsma, H.; Bombardieri, M. Blocking T cell co-stimulation in primary Sjögren’s syndrome: Rationale, clinical efficacy and modulation of peripheral and salivary gland biomarkers. Clin. Exp. Rheumatol 2020, 126, 222–227. [Google Scholar]
- Van Nimwegen, J.F.; Mossel, E.; van Zuiden, G.S.; Wijnsma, R.F.; Delli, K.; Stel, A.J.; van der Vegt, B.; Haacke, E.A.; Olie, L.; Los, L.I.; et al. Abatacept treatment for patients with early active primary Sjögren’s syndrome: A single-centre, randomised, double-blind, placebo-controlled, phase 3 trial (ASAP-III study). Lancet Rheumatol. 2020, 2, E153–E163. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Meiners, P.M.; Corneth, O.B.J.; Visser, A.; Arends, S.; Abdulahad, W.H.; Hendriks, R.W.; Vissink, A.; Kroese, F.G.M.; Bootsma, H. Attenuation of follicular helper T cell-dependent B Cell hyperactivity by abatacept treatment in primary Sjögren’s syndrome. Arthritis Rheumatol. 2017, 69, 1850–1861. [Google Scholar] [CrossRef]
- Hoogen, L.L.V.D.; van Laar, J.M. Targeted therapies in systemic sclerosis, myositis, antiphospholipid syndrome, and Sjögren’s syndrome. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101485. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT02843659 (accessed on 15 January 2021).
- Van Berkel, M.E.; Oosterwegel, M.A. CD28 and ICOS: Similar or separate costimulators of T cells? Immunol. Lett. 2006, 105, 115–122. [Google Scholar] [CrossRef]
- Bombardieri, M.; Alevizos, I.; Moate, R.; Sullivan, B.; Noaiseh, G.; Kvarnström, M.; Rees, W.; Wang, L.; Illei, G. Phase 2a study of MEDI5872 (AMG557), a fully human anti-ICOS ligand monoclonal antibody in patients with primary Sjögren’s syndrome. Arthritis Rheumatol. 2019, 71 (Suppl. 10), 2417. [Google Scholar]
- Generali, E.; Bose, T.; Selmi, C.; Voncken, J.W.; Damoiseaux, J. Nature versus nurture in the spectrum of rheumatic diseases: Classification of spondyloarthritis as autoimmune or autoinflammatory. Autoimmun. Rev. 2018, 17, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Belkhir, R.; Gestermann, N.; Koutero, M.; Seror, R.; Tost, J.; Mariette, X.; Richard-Miceli, C. Upregulation of membrane-bound CD40L on CD4+T cells in women with primary Sjögren’s syndrome. Scand. J. Immunol. 2013, 79, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.A.; Szanto, A.; Ng, W.-F.; Bombardieri, M.; Posch, M.G.; Papas, A.S.; Farag, A.M.; Daikeler, T.; Bannert, B.; Kyburz, D.; et al. Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: A multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol. 2020, 2, E142–E152. [Google Scholar] [CrossRef]
- De Souza, R.G.; Yu, Z.; Stern, M.E.; Pflugfelder, S.C.; De Paiva, C.S. Suppression of Th1-mediated keratoconjunctivitis sicca by lifitegrast. J. Ocul. Pharmacol. Ther. 2018, 34, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Waickman, A.T.; Park, J.-Y.; Park, J.-H. The common γ-chain cytokine receptor: Tricks-and-treats for T cells. Cell. Mol. Life Sci. 2016, 73, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gao, H.; Wang, Q.; Wang, M.; Wu, B. Molecular mechanisms and clinical application of Iguratimod: A review. Biomed. Pharmacother. 2020, 122, 109704. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, L.; Zhao, Y.; He, X.; Hu, C.; Liu, Y. The efficacy and mechanism for action of iguratimod in primary Sjögren’s syndrome patients. Int. Ophthalmol. 2020, 40, 3059–3065. [Google Scholar] [CrossRef]
- Shao, Q.; Wang, S.; Jiang, H.; Liu, L. Efficacy and safety of iguratimod on patients with primary Sjögren’s syndrome: A randomized, placebo-controlled clinical trial. Scand. J. Rheumatol. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Roescher, N.; Tak, P.P.; Illei, G.G. Cytokines in Sjögren’s syndrome. Oral Dis. 2009, 15, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Sankar, V.; Brennan, M.T.; Kok, M.R.; Leakan, R.A.; Smith, J.A.; Manny, J.; Baum, B.J.; Pillemer, S.R. Etanercept in Sjögren’s syndrome: A twelve-week randomized, double-blind, placebo-controlled pilot clinical trial. Arthritis Rheum. 2004, 50, 2240–2245. [Google Scholar] [CrossRef]
- Mariette, X.; Ravaud, P.P.; Steinfeld, S.; Baron, G.; Goetz, J.; Hachulla, E.; Combe, B.; Puéchal, X.; Pennec, Y.Y.; Sauvezie, B.B.; et al. Inefficacy of infliximab in primary Sjögren’s syndrome: Results of the randomized, controlled trial of remicade in primary Sjögren’s syndrome (TRIPSS). Arthritis Rheum. 2004, 50, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Norheim, K.B.; Harboe, E.; Gøransson, L.G.; Omdal, R. Interleukin-1 inhibition and fatigue in primary Sjögren’s syndrome—A double blind, randomised clinical trial. PLoS ONE 2012, 7, e30123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komai, T.; Shoda, H.; Yamaguchi, K.; Sakurai, K.; Shibuya, M.; Kubo, K.; Takahashi, T.; Fujio, K.; Yamamoto, K. Neuromyelitis optica spectrum disorder complicated with Sjögren syndrome successfully treated with tocilizumab: A case report. Mod. Rheumatol. 2013, 26, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, J.; Hattori, K.; Fujiwara, M.; Kita, Y. Refractory Sjögren’s syndrome myelopathy successfully treated with subcutaneous tocilizumab. Medicine 2019, 98, e16285. [Google Scholar] [CrossRef] [PubMed]
- Mosca, P.J.; Clay, T.M.; Morse, M.A.; Lyerly, H.K. Immune monitoring. Cancer Treat. Res. 2005, 123, 369–388. [Google Scholar]
- Abdulahad, W.H.; Meijer, J.M.; Kroese, F.G.M.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.; Kallenberg, C.G.M.; Bootsma, H. B cell reconstitution and T helper cell balance after rituximab treatment of active primary Sjögren’s syndrome: A double-blind, placebo-controlled study. Arthritis Rheum. 2011, 63, 1116–1123. [Google Scholar] [CrossRef]
- Gatti, A.; Buccisano, F.; Scupoli, M.T.; Brando, B. The ISCCA flow protocol for the monitoring of anti-CD20 therapies in autoimmune disorders. Cytometry B Clin. Cytom. 2020. [Google Scholar] [CrossRef]
- Thiel, J.; Rizzi, M.; Engesser, M.; Dufner, A.-K.; Troilo, A.; Lorenzetti, R.; Voll, R.E.; Venhoff, N. B cell repopulation kinetics after rituximab treatment in ANCA-associated vasculitides compared to rheumatoid arthritis, and connective tissue diseases: A longitudinal observational study on 120 patients. Arthritis Res. 2017, 19, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Anolik, J.H.; Campbell, D.; Felgar, R.E.; Young, F.; Sanz, I.; Rosenblatt, J.; Looney, R.J. The relationship of FcγRIIIa genotype to degree of B cell depletion by rituximab in the treatment of systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 455–459. [Google Scholar] [CrossRef]
- Pers, J.-O.; Daridon, C.; Bendaoud, B.; Devauchelle, V.; Berthou, C.; Saraux, A.; Youinou, P. B-cell depletion and repopulation in autoimmune diseases. Clin. Rev. Allergy Immunol. 2007, 34, 50–55. [Google Scholar] [CrossRef]
- Boyer-Suavet, S.; Andreani, M.; Lateb, M.; Savenkoff, B.; Brglez, V.; Benzaken, S.; Bernard, G.; Nachman, P.H.; Esnault, V.; Seitz-Polski, B. Neutralizing anti-rituximab antibodies and relapse in membranous nephropathy treated with rituximab. Front. Immunol. 2020, 10, 3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrion, C.; Guérin, E.; Gachard, N.; Le Guyader, A.; Giraut, S.; Feuillard, J. Adult bone marrow three-dimensional phenotypic landscape of B-cell differentiation. Cytom. Part B Clin. Cytom. 2019, 96, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binard, A.; Le Pottier, L.; Devauchelle-Pensec, V.; Saraux, A.; Youinou, P.; Pers, J.-O. Is the blood B-cell subset profile diagnostic for Sjögren syndrome? Ann. Rheum. Dis. 2008, 68, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Roll, P.; Palanichamy, A.; Kneitz, C.; Dorner, T.; Tony, H.-P. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Leandro, M.; Cambridge, G.; Ehrenstein, M.R.; Edwards, J.C.W. Reconstitution of peripheral blood B cells after depletion with rituximab in patients with rheumatoid arthritis. Arthritis Rheum. 2006, 54, 613–620. [Google Scholar] [CrossRef]
- Ramsköld, D.; Parodis, I.; Lakshmikanth, T.; Sippl, N.; Khademi, M.; Chen, Y.; Zickert, A.; Mikeš, J.; Achour, A.; Amara, K.; et al. B cell alterations during BAFF inhibition with belimumab in SLE. EBioMedicine 2019, 40, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Gensous, N.; Charrier, M.; Duluc, D.; Contin-Bordes, C.; Truchetet, M.-E.; Lazaro, E.; Duffau, P.; Blanco, P.; Richez, C. T follicular helper cells in autoimmune disorders. Front. Immunol. 2018, 9, 1637. [Google Scholar] [CrossRef]
- Kurata, I.; Matsumoto, I.; Sumida, T. T follicular helper cell subsets: A potential key player in autoimmunity. Immunol. Med. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Dionne, K.; Redfern, R.L.; Nichols, J.J.; Nichols, K.K. Analysis of tear inflammatory mediators: A comparison between the microarray and Luminex methods. Mol. Vis. 2016, 22, 177–188. [Google Scholar]
- Zhao, H.; Li, Q.; Ye, M.; Yu, J. Tear Luminex analysis in dry eye patients. Med. Sci. Monit. 2018, 24, 7595–7602. [Google Scholar] [CrossRef]
- Chen, X.; Aqrawi, L.A.; Utheim, T.P.; Tashbayev, B.; Utheim, Ø.A.; Reppe, S.; Hove, L.H.; Herlofson, B.B.; Singh, P.B.; Palm, Ø.; et al. Elevated cytokine levels in tears and saliva of patients with primary Sjögren’s syndrome correlate with clinical ocular and oral manifestations. Sci. Rep. 2019, 9, 7319. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.T.; Lindstrom, T.M.; Robinson, W.H.; Utz, P.J.; Hale, M.B.; Boyd, S.D.; Shen-Orr, S.S.; Fathman, C.G. New tools for classification and monitoring of autoimmune diseases. Nat. Rev. Rheumatol. 2012, 8, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, F.J.; Bendall, S. Immune monitoring using mass cytometry and related high-dimensional imaging approaches. Nat. Rev. Rheumatol. 2019, 16, 87–99. [Google Scholar] [CrossRef]
- Hartmann, F.J.; Babdor, J.; Gherardini, P.F.; Amir, E.-A.D.; Jones, K.; Sahaf, B.; Marquez, D.M.; Krutzik, P.; O’Donnell, E.; Sigal, N.; et al. Comprehensive immune monitoring of clinical trials to advance human immunotherapy. Cell Rep. 2019, 28, 819–831.e4. [Google Scholar] [CrossRef] [Green Version]
- Marco, H.; Smith, R.M.; Jones, R.B.; Guerry, M.-J.; Catapano, F.; Burns, S.; Chaudhry, A.N.; Smith, K.G.C.; Jayne, D.R.W. The effect of rituximab therapy on immunoglobulin levels in patients with multisystem autoimmune disease. BMC Musculoskelet. Disord. 2014, 15, 178. [Google Scholar] [CrossRef]
- Reff, M.E.; Carner, K.; Chambers, K.S.; Chinn, P.C.; Leonard, J.E.; Raab, E.; Newman, R.A.; Hanna, N.; Andreson, D.R. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994, 83, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Kamburova, E.G.; Koenen, H.J.P.M.; Borgman, K.J.E.; ten Berge, I.J.; Joosten, I.; Hilbrands, L.B. A single dose of rituximab does not deplete B Cells in secondary lymphoid organs but alters phenotype and function. Am. J. Transplant. 2013, 13, 1503–1511. [Google Scholar] [CrossRef]
- Gea-Banacloche, J. Rituximab-associated infections. Semin. Hematol. 2010, 47, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.-H.; Hwang, W.-L.; Cheng, S.-B.; Lee, T.-Y.; Teng, C.-L. Hepatitis B reactivation induced by Rituximab maintenance therapy for lymphoma. Ann. Hematol. 2010, 90, 111–112. [Google Scholar] [CrossRef]
- Oren, S.; Mandelboim, M.; Braunmoscovici, Y.; Paran, D.; Ablin, J.N.; Litinsky, I.; Comaneshter, D.; Levartovsky, D.; Mendelson, E.; Azar, R.; et al. Vaccination against influenza in patients with rheumatoid arthritis: The effect of rituximab on the humoral response. Ann. Rheum. Dis. 2008, 67, 937–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehnberg, M.; Brisslert, M.; Amu, S.; Zendjanchi, K.; Håwi, G.; Bokarewa, M.I. Vaccination response to protein and carbohydrate antigens in patients with rheumatoid arthritis after rituximab treatment. Arthritis Res. Ther. 2010, 12, R111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Assen, S.; Holvast, A.; Benne, C.A.; Posthumus, M.D.; van Leeuwen, M.A.; Voskuyl, A.E.; Blom, M.; Risselada, A.P.; de Haan, A.; Westra, J.; et al. Humoral responses after influenza vaccination are severely reduced in patients with rheumatoid arthritis treated with rituximab. Arthritis Rheum. 2009, 62, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, M.; Kalina, T.; Perez-Andres, M.; Vlkova, M.; Lopez-Granados, E.; Blanco, E.; Bonroy, C.; Sousa, A.E.; Kienzler, A.-K.; Wentink, M.; et al. The EuroFlow PID orientation tube for flow cytometric diagnostic screening of primary immunodeficiencies of the lymphoid system. Front. Immunol. 2019, 10, 246. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, J.J.; van der Burg, M.; Kalina, T.; Perez-Andres, M.; Mejstrikova, E.; Vlkova, M.; Lopez-Granados, E.; Wentink, M.; Kienzler, A.-K.; Philippé, J.; et al. EuroFlow-based flowcytometric diagnostic screening and classification of primary immunodeficiencies of the lymphoid system. Front. Immunol. 2019, 10, 1271. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Item | Weight/Score | Rules for Classification |
|---|---|---|
| 1. Labial salivary gland with focal lymphocytic sialadenitis and focus score of ≥ 1 foci/4 mm2 | 3 | Applies to any individual |
| 2. Anti-SSA/Ro-positive | 3 | —who meets the inclusion criteria (presence of ocular and/or oral dryness) with at least one symptom of ocular or oral dryness or ESSDAI ≥ 1 |
| 3. Ocular Staining Score ≥ 5 (or van Bijsterveld score ≥ 4) in at least one eye | 1 | —does not have any of the conditions listed as exclusion criteria a |
| 4. Schirmer’s test ≤ 5 mm/5 min in at least one eye | 1 | —and has a score of ≥ 4 when the weights from the 5 criteria items are summed |
| 5. Unstimulated whole saliva flow rate ≤ 0.1 mL/min | 1 |
| Timepoint | Suggested Assay |
|---|---|
| Baseline a | IgG, IgM, IgA concentrations, B-cell numbers including memory B-cell subsets (un-switched and switched; IgD+/CD27+/CD38- and IgD-/CD27+/CD38-, respectively) and plasmablast (IgD-/CD27+/CD38++) numbers. Optional (depending on therapy): cytokine concentrations b |
| 3 months after therapy onset c | B-cell numbers including memory B-cell subsets (un-switched and switched; IgD+/CD27+/CD38- and IgD-/CD27+/CD38-, respectively) and plasmablast (IgD-/CD27+/CD38++) numbers. |
| 6–9 months after therapy onset | Lymphocyt (NK cells, T-cells and B-cells) numbers including memory B-cell subsets (un-switched and switched; IgD+/CD27+/CD38- and IgD-/CD27+/CD38-, respectively) and transitional B-cell (IgD+/CD27-/CD38++) numbers. Optional (depending on therapy): cytokine concentrations b |
| Infections | IgG, IgM, IgA concentrations, T-cell numbers and B-cell numbers including memory B-cell subsets (un-switched and switched; IgD+/CD27+/CD38- and IgD-/CD27+/CD38-, respectively). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Beers, J.J.B.C.; Damoiseaux, J.G.M.C. Immune Monitoring upon Treatment with Biologics in Sjögren’s Syndrome: The What, Where, When, and How. Biomolecules 2021, 11, 116. https://doi.org/10.3390/biom11010116
van Beers JJBC, Damoiseaux JGMC. Immune Monitoring upon Treatment with Biologics in Sjögren’s Syndrome: The What, Where, When, and How. Biomolecules. 2021; 11(1):116. https://doi.org/10.3390/biom11010116
Chicago/Turabian Stylevan Beers, Joyce J.B.C., and Jan G.M.C. Damoiseaux. 2021. "Immune Monitoring upon Treatment with Biologics in Sjögren’s Syndrome: The What, Where, When, and How" Biomolecules 11, no. 1: 116. https://doi.org/10.3390/biom11010116
APA Stylevan Beers, J. J. B. C., & Damoiseaux, J. G. M. C. (2021). Immune Monitoring upon Treatment with Biologics in Sjögren’s Syndrome: The What, Where, When, and How. Biomolecules, 11(1), 116. https://doi.org/10.3390/biom11010116