
Cell-Projection Pumping of Fibroblast Contents into Osteosarcoma SAOS-2 Cells Correlates with Increased SAOS-2 Proliferation and Migration, as well as Altered Morphology

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Labelling of Cells with Lipophilic Fluorescent Membrane Markers
2.4. Co-Culture Conditions
2.5. Time-Lapse Recordings
2.6. Single-Cell Tracking and Analysis
2.7. Segmentation of Cells and Dependent Calculations for Cell Circularity, Absolute Fluorescence and Compensated Fluorescence
2.8. Determination of Cell Migration Velocity
2.9. Normalization of Fluorescence Values
2.10. Correlation by Kendall’s Tau of Cell-Profile Area, Cell Circularity and Cell Migration Velocity with Receipt of HDF Fluorescence
2.11. Correlation of Mitosis with Receipt of HDF Fluorescence
2.12. Calculation of an Index for Persistence of Phenotypic Effect Inherited from Mother Cells
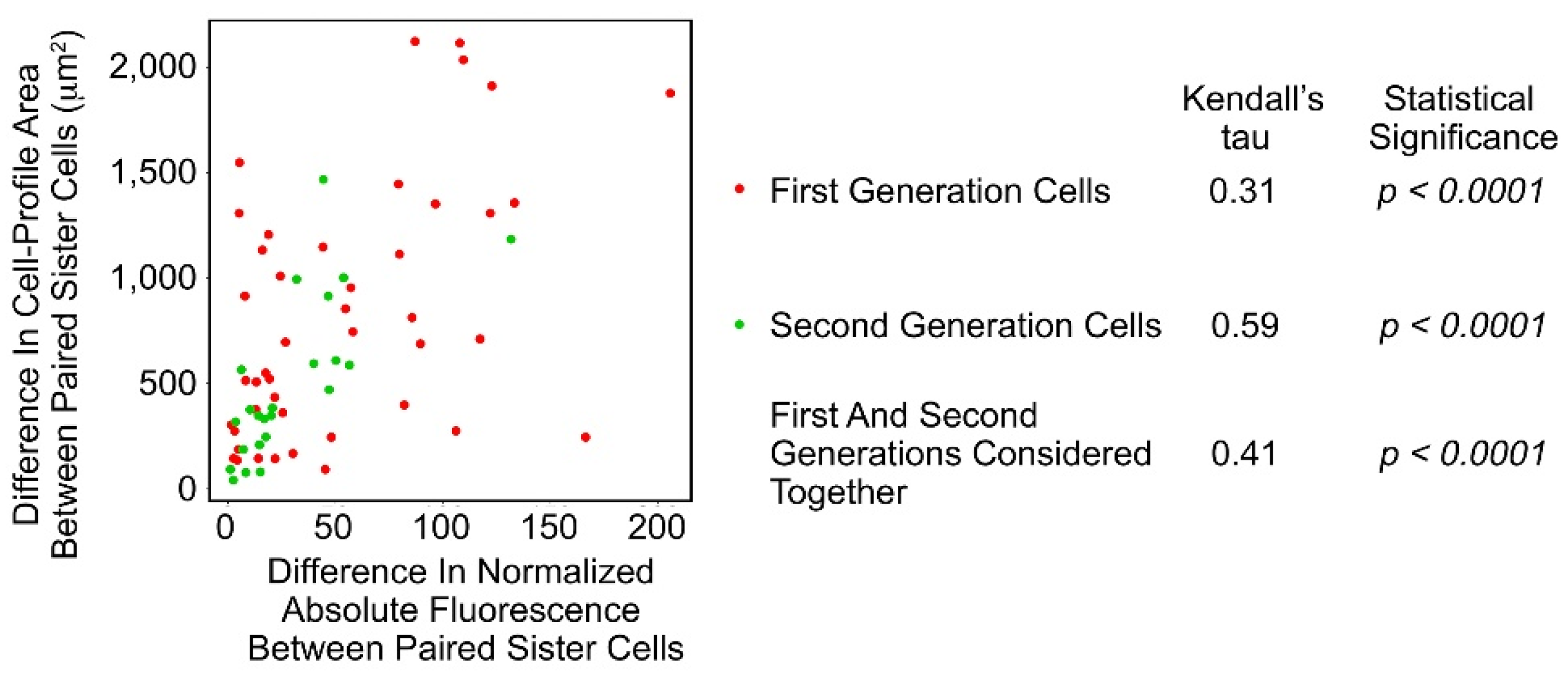
2.13. Analysis of the Relationship between Acquired Fluorescence and Phenotype in Paired SAOS-2 Sister Cells
2.14. Evaluation of Statistical Significance
3. Results
3.1. There Was Marked Transfer of HDF Fluorescent Label to SAOS-2 during Co-Culture
3.2. SAOS-2 Cells Had Lower Cell-Profile Area and Migration Velocity and Higher Cell Circularity than HDF
3.3. SAOS-2 Cell-Profile Area Correlated with Receipt of HDF Fluorescence and the Effect Did Not Persist Post-Mitosis
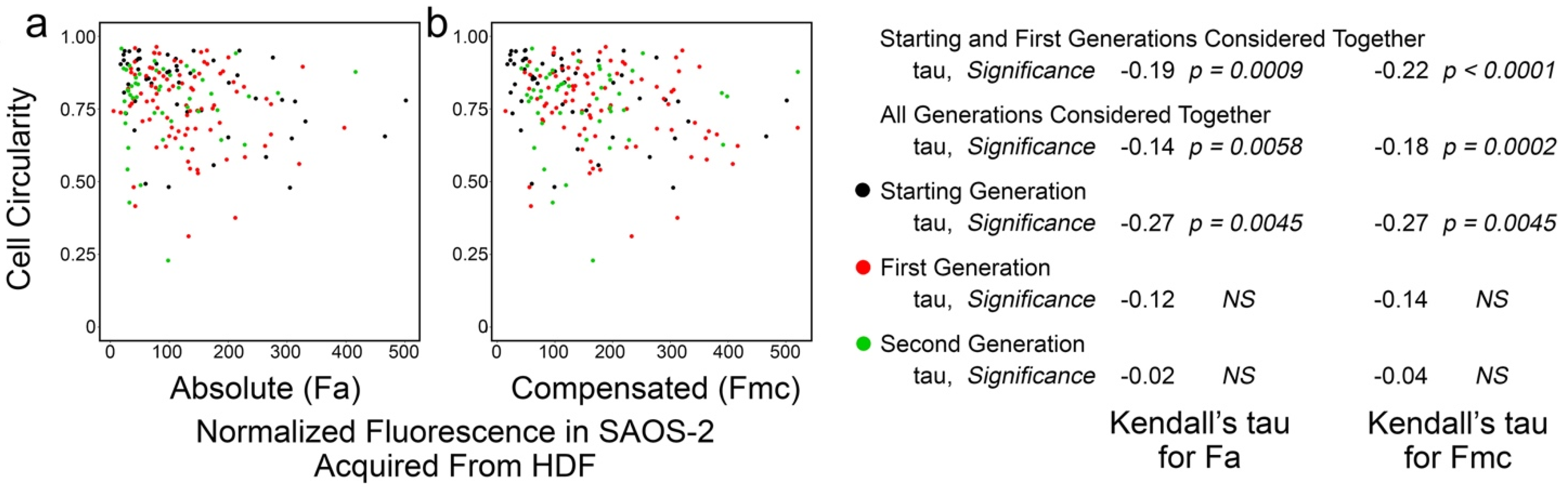
3.4. Cell Circularity in Co-Cultured SAOS-2 Cells Was Inversely Correlated with Receipt of HDF Fluorescence and the Effect Persisted Post-Mitosis
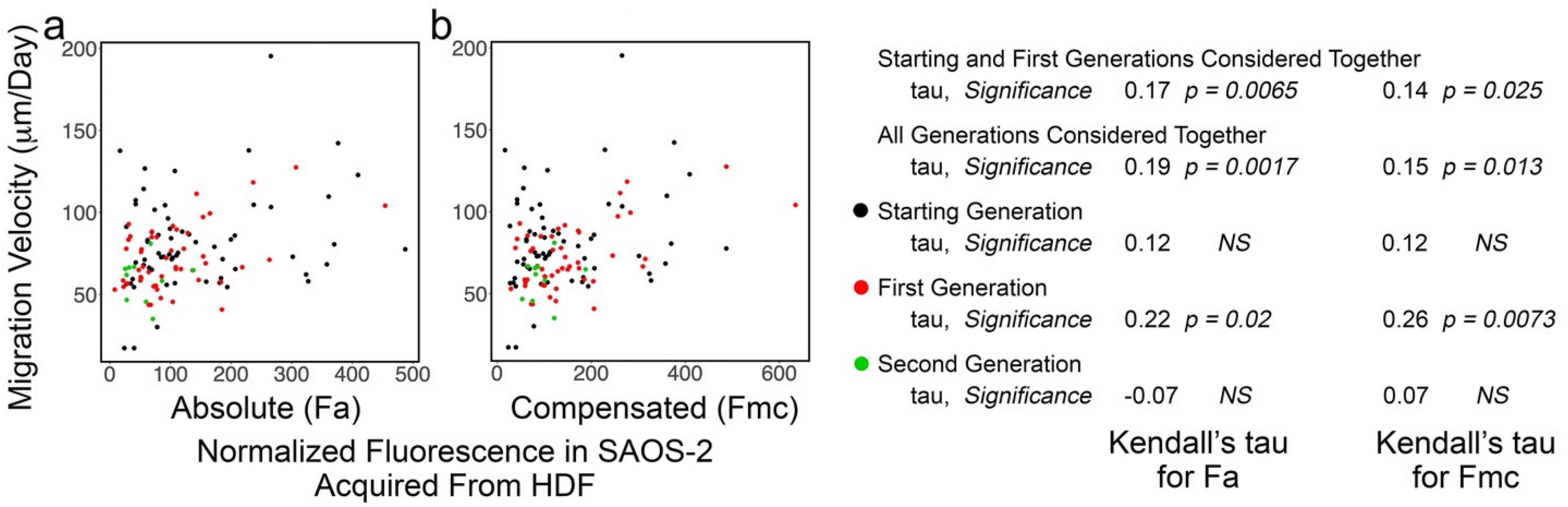
3.5. SAOS-2 Cell Migration Velocity Had Weak Correlation with Receipt of HDF Fluorescence and the Effect Did Not Persist Post-SAOS-2 Cell Division
3.6. Increased HDF Fluorescence Transfer to SAOS-2 Cells during Co-Culture Was Associated with Subsequent SAOS-2 Cell Mitosis and There Was No Evidence of Persistence of This Post-Cell Division
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- David, M.S.; Huynh, M.D.; Kelly, E.; Rizos, H.; Coleman, H.; Rogers, G.; Zoellner, H. Membrane and cytoplasmic marker exchange between malignant neoplastic cells and fibroblasts via intermittent contact: Increased tumour cell diversity independent of genetic change. J. Pathol. 2012, 228, 495–505. [Google Scholar] [CrossRef]
- Zoellner, H.; Chami, B.; Kelly, E.; Moore, M.A.S. Increased cell size, structural complexity and migration of cancer cells acquiring fibroblast organelles by cell-projection pumping. PLoS ONE 2019, 14, e0224800. [Google Scholar] [CrossRef] [Green Version]
- Zoellner, H.; Paknejad, N.; Cornwell, J.A.; Chami, B.; Romin, Y.; Boyko, V.; Fujisawa, S.; Kelly, E.; Lynch, G.W.; Rogers, G.; et al. Potential Hydrodynamic Cytoplasmic Transfer between Mammalian Cells: Cell-Projection Pumping. Biophys. J. 2020, 118, 1248–1260. [Google Scholar] [CrossRef]
- Koyanagi, M.; Brandes, R.P.; Haendeler, J.; Zeiher, A.M.; Dimmeler, S. Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: A novel mechanism for cell fate changes? Circ. Res. 2005, 96, 1039–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, K.A.; Yerkovich, S.T.; Hopkins, P.M.; Chambers, D.C. Characterization of intercellular communication and mitochondrial donation by mesenchymal stromal cells derived from the human lung. Stem Cell Res. Ther. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhaneni, K.C.; Haller, H.; Dumler, I. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2012, 21, 3104–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, D.M.; Sowinski, S. Membrane nanotubes: Dynamic long-distance connections between animal cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 431–436. [Google Scholar] [CrossRef]
- Gerdes, H.H.; Rustom, A.; Wang, X. Tunneling nanotubes, an emerging intercellular communication route in development. Mech. Dev. 2013, 130, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Onfelt, B.; Nedvetzki, S.; Benninger, R.K.; Purbhoo, M.A.; Sowinski, S.; Hume, A.N.; Seabra, M.C.; Neil, M.A.; French, P.M.; Davis, D.M. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guescini, M.; Leo, G.; Genedani, S.; Carone, C.; Pederzoli, F.; Ciruela, F.; Guidolin, D.; Stocchi, V.; Mantuano, M.; Borroto-Escuela, D.O.; et al. Microvesicle and tunneling nanotube mediated intercellular transfer of g-protein coupled receptors in cell cultures. Exp. Cell Res. 2012, 318, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Thayanithy, V.; Babatunde, V.; Dickson, E.L.; Wong, P.; Oh, S.; Ke, X.; Barlas, A.; Fujisawa, S.; Romin, Y.; Moreira, A.L.; et al. Tumor exosomes induce tunneling nanotubes in lipid raft-enriched regions of human mesothelioma cells. Exp. Cell Res. 2014, 323, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, E.; Fujisawa, S.; Barlas, A.; Romin, Y.; Manova-Todorova, K.; Moore, M.A.; Subramanian, S. Tunneling Nanotubes: A new paradigm for studying intercellular communication and therapeutics in cancer. Commun. Integr. Biol. 2012, 5, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Ariazi, J.; Benowitz, A.; De Biasi, V.; Den Boer, M.L.; Cherqui, S.; Cui, H.; Douillet, N.; Eugenin, E.A.; Favre, D.; Goodman, S.; et al. Tunneling Nanotubes and Gap Junctions-Their Role in Long-Range Intercellular Communication during Development, Health, and Disease Conditions. Front. Mol. Neurosci. 2017, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; Pan, H.; Lanza, G.M.; Wickline, S.A. Consortium for Translational Research in Advanced Imaging and Nanomedicine (C-TRAIN). Paracrine induction of endothelium by tumor exosomes. Lab. Invest. 2009, 89, 1317–1328. [Google Scholar] [PubMed] [Green Version]
- Wu, H.H.; Lee, O.K. Exosomes from mesenchymal stem cells induce the conversion of hepatocytes into progenitor oval cells. Stem Cell Res. Ther. 2017, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- McCready, J.; Sims, J.D.; Chan, D.; Jay, D.G. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: A role for plasminogen activation. BMC Cancer 2010, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, S.J.; McCoy-Simandle, K.; Leung, E.; Genna, A.; Condeelis, J.; Cox, D. Tunneling nanotubes, a novel mode of tumor cell-macrophage communication in tumor cell invasion. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res. 2014, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Matula, Z.; Mikala, G.; Lukacsi, S.; Matko, J.; Kovacs, T.; Monostori, E.; Uher, F.; Valyi-Nagy, I. Stromal Cells Serve Drug Resistance for Multiple Myeloma via Mitochondrial Transfer: A Study on Primary Myeloma and Stromal Cells. Cancers 2021, 13, 3461. [Google Scholar] [CrossRef]
- Caicedo, A.; Fritz, V.; Brondello, J.M.; Ayala, M.; Dennemont, I.; Abdellaoui, N.; de Fraipont, F.; Moisan, A.; Prouteau, C.A.; Boukhaddaoui, H.; et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 2015, 5, 9073. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Wang, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [Green Version]
- Salaud, C.; Alvarez-Arenas, A.; Geraldo, F.; Belmonte-Beitia, J.; Calvo, G.F.; Gratas, C.; Pecqueur, C.; Garnier, D.; Perez-Garcia, V.; Vallette, F.M.; et al. Mitochondria transfer from tumor-activated stromal cells (TASC) to primary Glioblastoma cells. Biochem. Biophys. Res. Commun. 2020, 533, 139–147. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Zampieri, L.X.; Silva-Almeida, C.; Rondeau, J.D.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.A.; Xiang, J. Mechanisms of cellular communication through intercellular protein transfer. J. Cell Mol. Med. 2011, 15, 1458–1473. [Google Scholar] [CrossRef]
- Ady, J.W.; Desir, S.; Thayanithy, V.; Vogel, R.I.; Moreira, A.L.; Downey, R.J.; Fong, Y.; Manova-Todorova, K.; Moore, M.A.; Lou, E. Intercellular communication in malignant pleural mesothelioma: Properties of tunneling nanotubes. Front. Physiol. 2014, 5, 400. [Google Scholar] [CrossRef]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.S.; Kelly, E.; Zoellner, H. Opposite cytokine synthesis by fibroblasts in contact co-culture with osteosarcoma cells compared with transwell co-cultures. Cytokine 2013, 62, 48–51. [Google Scholar] [CrossRef]
- Kumar, V.; Abbas, A.K. Robbins and Cotran Pathologic Basis of Disease, 9th ed.; Elsevier/Saunders: Philadelphia, PA, USA, 2014. [Google Scholar]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Han, Y.; Cho, U.; Kim, S.; Park, I.S.; Cho, J.H.; Dhanasekaran, D.N.; Song, Y.S. Tumour microenvironment on mitochondrial dynamics and chemoresistance in cancer. Free Radic. Res. 2018, 52, 1271–1287. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, M.S.; Kelly, E.; Cheung, I.; Xaymardan, M.; Moore, M.A.; Zoellner, H. SAOS-2 osteosarcoma cells bind fibroblasts via ICAM-1 and this is increased by tumour necrosis factor-alpha. PLoS ONE 2014, 9, e101202. [Google Scholar] [CrossRef] [Green Version]
- Krakhmal, N.V.; Zavyalova, M.V.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V.M. Cancer Invasion: Patterns and Mechanisms. Acta. Naturae 2015, 7, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornwell, J.A.; Gutierrez, M.Z.; Harvey, R.P.; Nordon, R.E. Live cell imaging and single cell tracking of mesenchymal stromal cells in vitro. In The Biology and Therapeutic Application of Mesenchymal Cells; Atkinson, K., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 326–346. [Google Scholar]
- Cornwell, J.A.; Mahadevan, S.; Draper, J.S.; Joun, G.L.; Zoellner, H.; Asli, N.S.; Harvey, R.P.; Nordon, R.E. TrackPad: Software for semi-automated single-cell tracking and lineage annotation. SoftwareX 2020, 11, 1–7. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Paek, A.L.; Reyes, J.; Lasick, K.A.; Lahav, G.; Michor, F. Hidden heterogeneity and circadian-controlled cell fate inferred from single cell lineages. Nat. Commun. 2018, 9, 5372. [Google Scholar] [CrossRef]
- Edelstein, A.D.; Tsuchida, M.A.; Amodaj, N.; Pinkard, H.; Vale, R.D.; Stuurman, N. Advanced methods of microscope control using muManager software. J. Biol. Methods 2014, 1, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. 2020. Available online: https://www.R-project.org/ (accessed on 15 February 2021).
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Vodermaier, H.C. APC/C and SCF: Controlling each other and the cell cycle. Curr. Biol. 2004, 14, R787–R796. [Google Scholar] [CrossRef] [Green Version]
- Trcek, T.; Larson, D.R.; Moldon, A.; Query, C.C.; Singer, R.H. Single-molecule mRNA decay measurements reveal promoter- regulated mRNA stability in yeast. Cell 2011, 147, 1484–1497. [Google Scholar] [CrossRef] [Green Version]
- Eser, P.; Demel, C.; Maier, K.C.; Schwalb, B.; Pirkl, N.; Martin, D.E.; Cramer, P.; Tresch, A. Periodic mRNA synthesis and degradation co-operate during cell cycle gene expression. Mol. Syst. Biol. 2014, 10, 717. [Google Scholar] [CrossRef] [PubMed]
- Rambout, X.; Detiffe, C.; Bruyr, J.; Mariavelle, E.; Cherkaoui, M.; Brohee, S.; Demoitie, P.; Lebrun, M.; Soin, R.; Lesage, B.; et al. The transcription factor ERG recruits CCR4-NOT to control mRNA decay and mitotic progression. Nat. Struct. Mol. Biol. 2016, 23, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Battich, N.; Beumer, J.; de Barbanson, B.; Krenning, L.; Baron, C.S.; Tanenbaum, M.E.; Clevers, H.; van Oudenaarden, A. Sequencing metabolically labeled transcripts in single cells reveals mRNA turnover strategies. Science 2020, 367, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Bernis, C.; Vigneron, S.; Labbe, J.C.; Lorca, T. The anaphase-promoting complex: A key factor in the regulation of cell cycle. Oncogene 2005, 24, 314–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.M. The anaphase-promoting complex: Proteolysis in mitosis and beyond. Mol. Cell 2002, 9, 931–943. [Google Scholar] [CrossRef]
- Krenning, L.; Sonneveld, S.; Tanenbaum, M.E. Time-resolved single-cell sequencing identifies multiple waves of mRNA decay during mitotic exit. BioRxiv 2021, 1–35. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkasalias, T.; Flaberg, E.; Kashuba, V.; Alexeyenko, A.; Pavlova, T.; Savchenko, A.; Szekely, L.; Klein, G.; Guven, H. Inhibition of tumor cell proliferation and motility by fibroblasts is both contact and soluble factor dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 17188–17193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Abbas, A.K.; Fausto, N.; Robbins, S.L. Robbins and Cotran Pathologic Basis of Disease; Elsevier Saunders: Philadelphia, PA, USA, 2005. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kendall’s Tau of Correlation between Cell-Profile Area and Receipt of HDF Fluorescence by SAOS-2 Cells | ||
|---|---|---|
| Fa | Fmc | |
| * Experiment a | ||
| Kendall’s tau of all Generations Considered Together | 0.55 | 0.53 |
| Statistical Significance of the Above | <0.0001 | <0.0001 |
| Experiment b | ||
| Kendall’s tau of all Generations Considered Together | 0.15 | 0.11 |
| Statistical Significance of the Above | <0.0001 | <0.0001 |
| Experiment c | ||
| Kendall’s tau of all Generations Considered Together | 0.76 | 0.68 |
| Statistical Significance of the Above | <0.0001 | <0.0001 |
| Experiment d | ||
| Kendall’s tau of all Generations Considered Together | 0.51 | 0.45 |
| Statistical Significance of the Above | <0.0001 | <0.0001 |
| Experiment e | ||
| Kendall’s tau of all Generations Considered Together | 0.13 | 0.08 |
| Statistical Significance of the Above | 0.001 | 0.04 |
| Experiment f | ||
| Kendall’s tau of all Generations Considered Together | 0.55 | 0.51 |
| Statistical Significance of the Above | <0.0001 | <0.0001 |
| Experiment g | ||
| Kendall’s tau of all Generations Considered Together | 0.54 | 0.41 |
| Statistical Significance of the Above | <0.0001 | <0.0001 |
| Kendall’s Tau of Correlation between Cell Circularity and Receipt of HDF Fluorescenceby SAOS-2 Cells | ||
|---|---|---|
| Fa | Fmc | |
| * Experiment a | ||
| Kendall’s tau of Starting and First Generations Considered Together | −0.19 | −0.22 |
| Statistical Significance of the Above | 0.0009 | <0.0001 |
| Experiment b | ||
| Kendall’s tau of Starting and First Generations Considered Together | −0.02 | −0.05 |
| Statistical Significance of the Above | NS | NS (0.063) |
| Experiment c | ||
| Kendall’s tau of Starting and First Generations Considered Together | −0.35 | −0.36 |
| Statistical Significance of the Above | <0.0001 | <0.0001 |
| Experiment d | ||
| Kendall’s tau of Starting and First Generations Considered Together | −0.24 | −0.23 |
| Statistical Significance of the Above | 0.0024 | 0.0032 |
| Experiment e | ||
| Kendall’s tau of Starting and First Generations Considered Together | 0.10 | 0.16 |
| Statistical Significance of the Above | NS (0.06) | 0.0026 |
| Experiment f | ||
| Kendall’s tau of Starting and First Generations Considered Together | −0.12 | −0.16 |
| Statistical Significance of the Above | 0.061 | 0.012 |
| Experiment g | ||
| Kendall’s tau of Starting and First Generations Considered Together | −0.22 | −0.18 |
| Statistical Significance of the Above | <0.0001 | 0.0003 |
| Kendall’s Tau of Correlation between Cell Migration Velocity and Receipt of HDF Fluorescence by SAOS-2 Cells | ||
|---|---|---|
| Fa | Fmc | |
| Experiment a | ||
| Kendall’s tau of Starting and First Generations Considered Together | 0.10 | 0.18 |
| Statistical Significance of the Above | NS (0.088) | 0.0019 |
| Experiment b | ||
| Kendall’s tau of Starting and First Generations Considered Together | 0.02 | −0.02 |
| Statistical Significance of the Above | NS | NS |
| Experiment c | ||
| Kendall’s tau of Starting and First Generations Considered Together | 0.25 | 0.19 |
| Statistical Significance of the Above | <0.0001 | <0.0002 |
| Experiment d | ||
| Kendall’s tau of Starting and First Generations Considered Together | 0.06 | 0.04 |
| Statistical Significance of the Above | NS | NS |
| Experiment e | ||
| Kendall’s tau of all Generations Considered Together | 0.08 | 0.06 |
| Statistical Significance of the Above | 0.045 | 0.11 |
| * Experiment f | ||
| Kendall’s tau of all Generations Considered Together | 0.19 | 0.15 |
| Statistical Significance of the Above | 0.0017 | 0.013 |
| Experiment g | ||
| Kendall’s tau of Starting, First and Second Generations Considered Together | 0.11 | 0.02 |
| Statistical Significance of the Above | 0.0069 | NS |
| Median Fa/Day | Median Fmc/Day | |||||
|---|---|---|---|---|---|---|
| Dividing Cells | Non-Dividing Cells | pFa (Dividing Cells/Non-Dividing Cells) | Dividing Cells | Non-Dividing Cells | pFmc (Dividing Cells/Non-Dividing Cells) | |
| Experiment a | 49.0 | 76.9 | 0.64 | 61.1 | 120.1 | 0.51 |
| Stat. Sig. of Above | 0.0022 | <0.0001 | ||||
| Experiment a* | 45.6 | 35.6 | 1.28 | 45.6 | 35.6 | - |
| Stat. Sig. of Above | NS | NS | ||||
| Experiment b | 89.4 | 73.3 | 1.22 | 113.5 | 153.3 | 0.74 |
| Stat. Sig. of Above | NS (0.057) | NS | ||||
| Experiment c | 66.7 | 29.9 | 2.23 | 51.4 | 11.4 | 4.51 |
| Stat. Sig. of Above | <0.0001 | <0.0001 | ||||
| Experiment d | 75.0 | 65.8 | 1.88 | 102.0 | 100.1 | 1.02 |
| Stat. Sig. of Above | NS (0.0535) | NS | ||||
| Experiment e | 39.9 | 15.8 | 2.53 | 73.0 | 50.0 | 1.46 |
| Stat. Sig. of Above | <0.0001 | 0.0041 | ||||
| Experiment f | 45.6 | 32.6 | 1.40 | 49.1 | 39.2 | 1.25 |
| Stat. Sig. of Above | 0.0222 | NS | ||||
| Experiment g | 64.9 | 42.1 | 1.54 | 94.0 | 65.9 | 1.43 |
| Stat. Sig. of Above | 0.0005 | 0.0115 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahadevan, S.; Cornwell, J.A.; Chami, B.; Kelly, E.; Zoellner, H. Cell-Projection Pumping of Fibroblast Contents into Osteosarcoma SAOS-2 Cells Correlates with Increased SAOS-2 Proliferation and Migration, as well as Altered Morphology. Biomolecules 2021, 11, 1875. https://doi.org/10.3390/biom11121875
Mahadevan S, Cornwell JA, Chami B, Kelly E, Zoellner H. Cell-Projection Pumping of Fibroblast Contents into Osteosarcoma SAOS-2 Cells Correlates with Increased SAOS-2 Proliferation and Migration, as well as Altered Morphology. Biomolecules. 2021; 11(12):1875. https://doi.org/10.3390/biom11121875
Chicago/Turabian StyleMahadevan, Swarna, James A Cornwell, Belal Chami, Elizabeth Kelly, and Hans Zoellner. 2021. "Cell-Projection Pumping of Fibroblast Contents into Osteosarcoma SAOS-2 Cells Correlates with Increased SAOS-2 Proliferation and Migration, as well as Altered Morphology" Biomolecules 11, no. 12: 1875. https://doi.org/10.3390/biom11121875
APA StyleMahadevan, S., Cornwell, J. A., Chami, B., Kelly, E., & Zoellner, H. (2021). Cell-Projection Pumping of Fibroblast Contents into Osteosarcoma SAOS-2 Cells Correlates with Increased SAOS-2 Proliferation and Migration, as well as Altered Morphology. Biomolecules, 11(12), 1875. https://doi.org/10.3390/biom11121875