
Glucagon Prevents Cytotoxicity Induced by Methylglyoxal in a Rat Neuronal Cell Line Model

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cytotoxicity Measurement
2.3. Cell Survival Assay
2.4. Apoptosis Estimation
2.5. Mitochondrial ROS Measurement
2.6. Neurite Outgrowth
2.7. cAMP Measurement
2.8. Protein Kinase A (PKA) Activity Detection
2.9. Statistical Analysis
3. Results
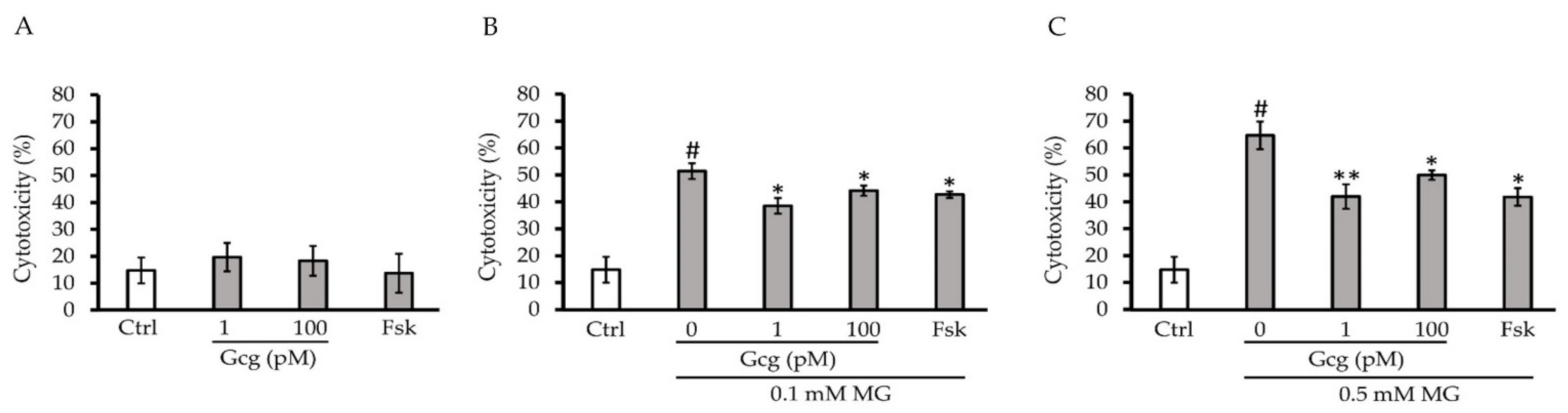
3.1. Glucagon Decreased Cytotoxicity Induced by MG
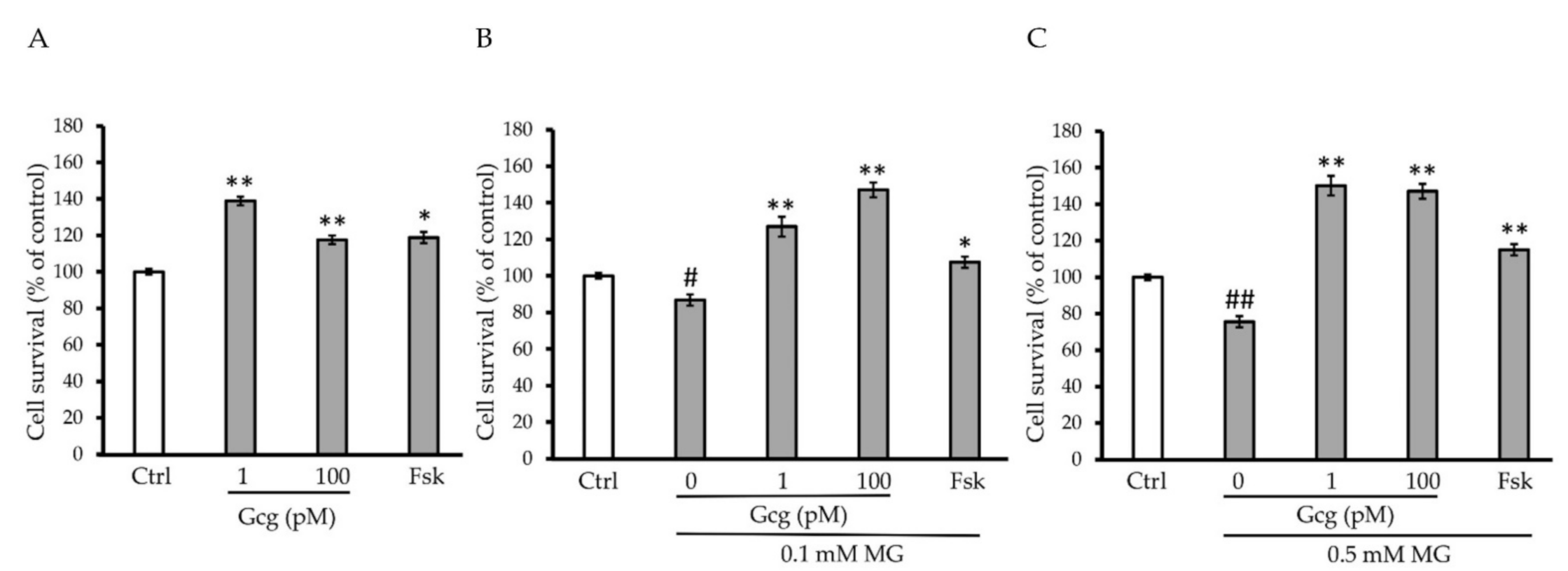
3.2. Survival of DRG Neuronal Cells Was Promoted by Glucagon
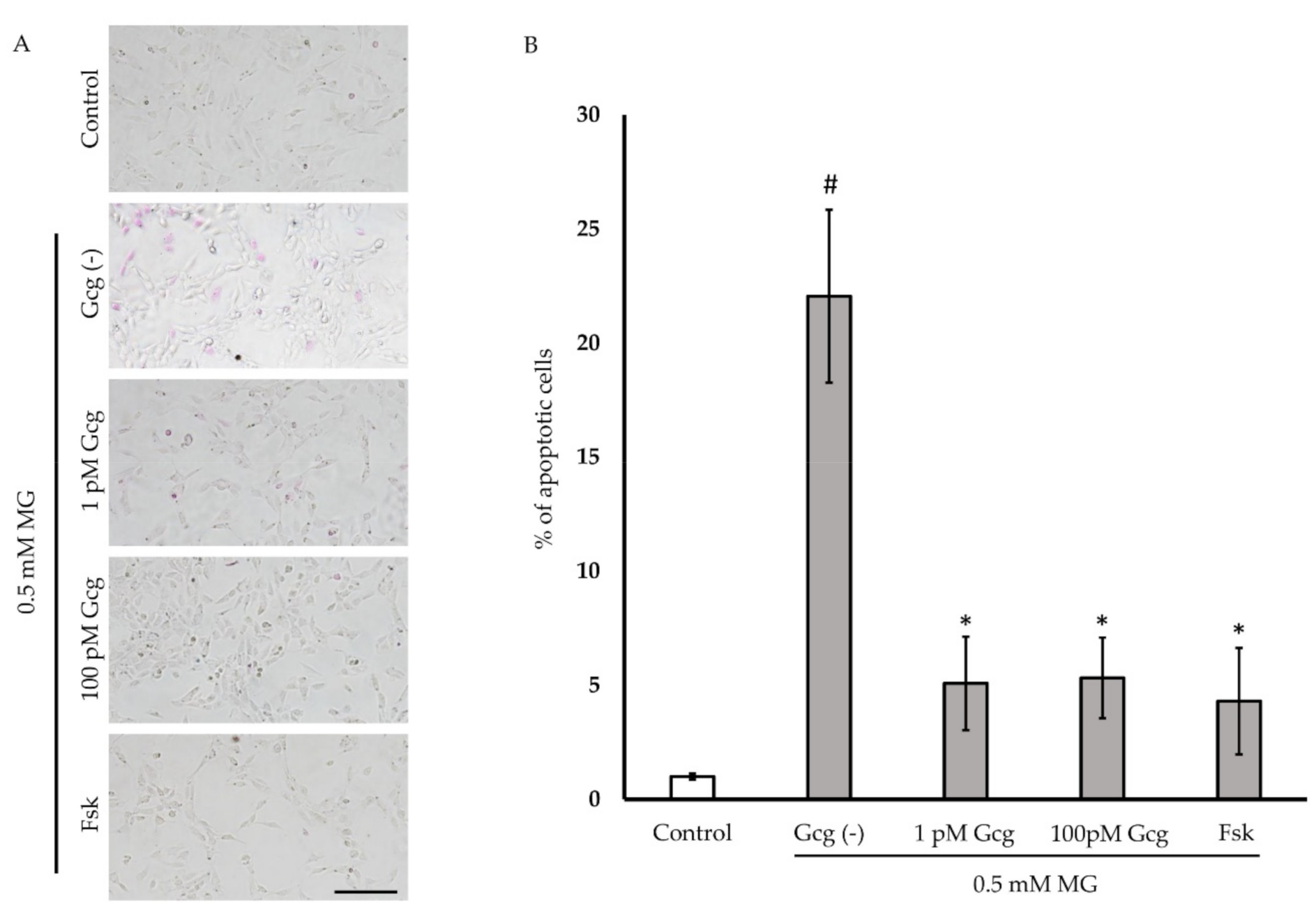
3.3. Glucagon Attenuated MG-Induced Apoptosis
3.4. Mitochondrial ROS Production Induced by MG Was Inhibited by Glucagon
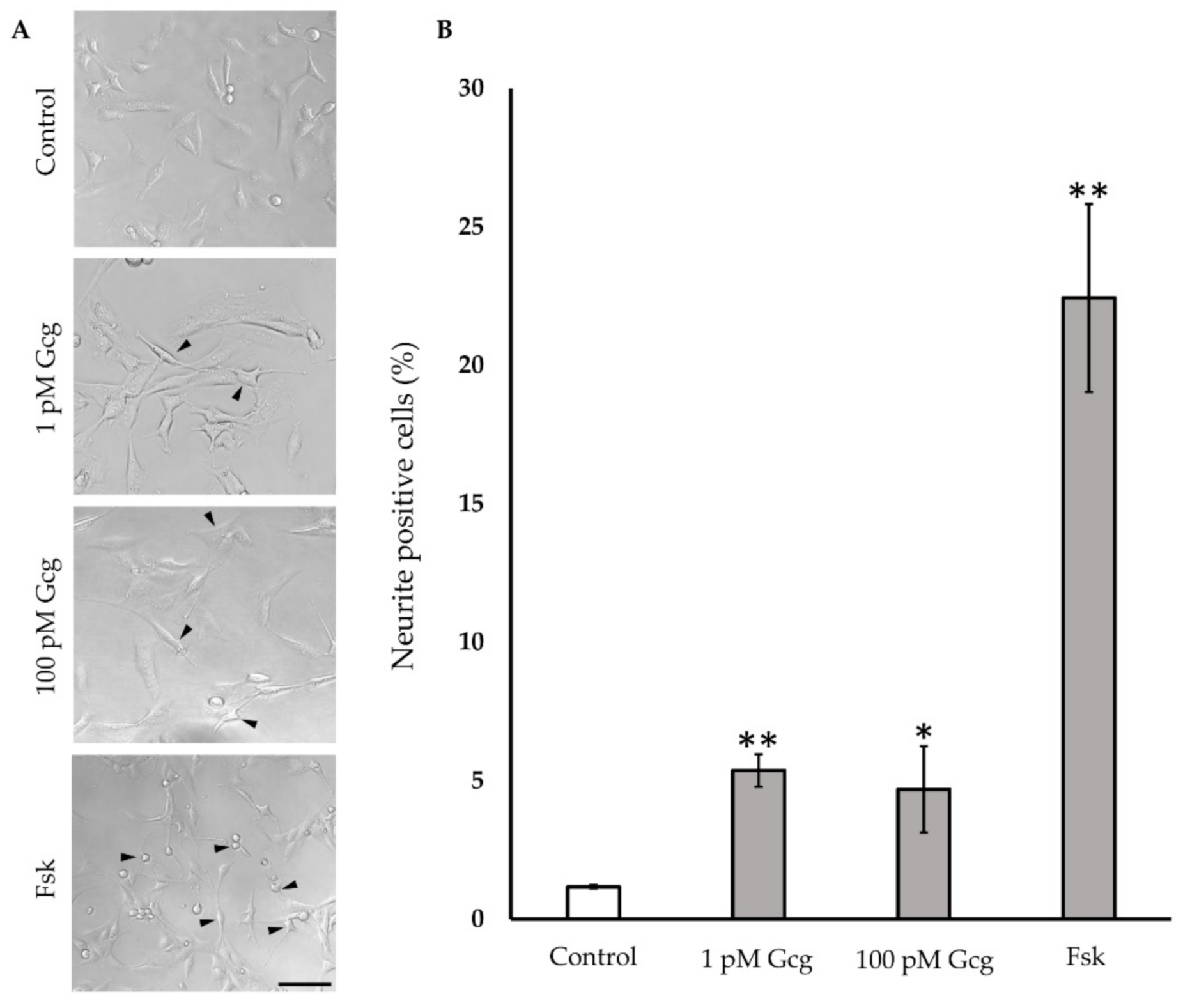
3.5. Neurite Projection Was Increased by Glucagon
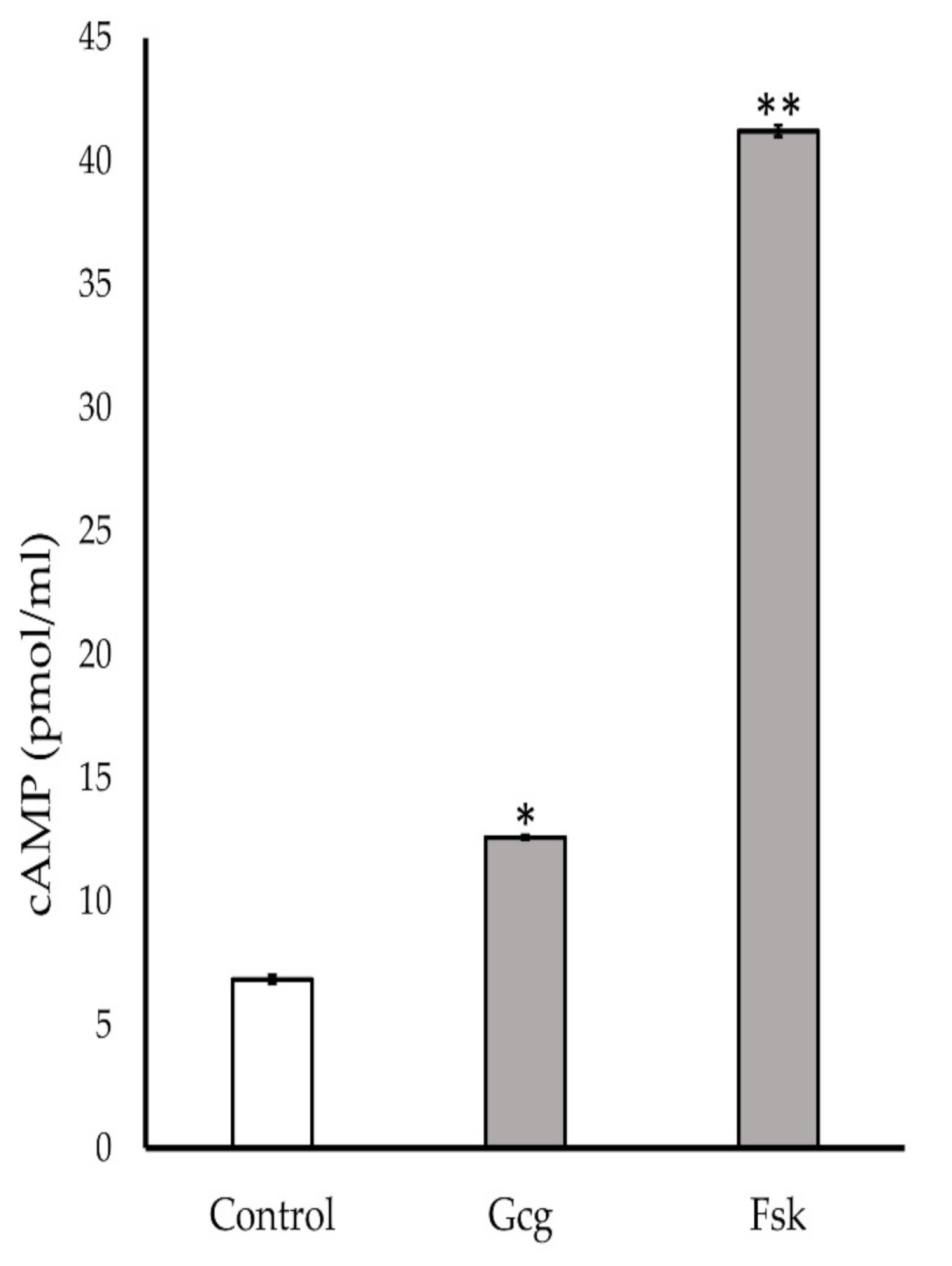
3.6. Production of Cyclic Adenosine Monophosphate (cAMP) Increased by Glucagon Stimulation
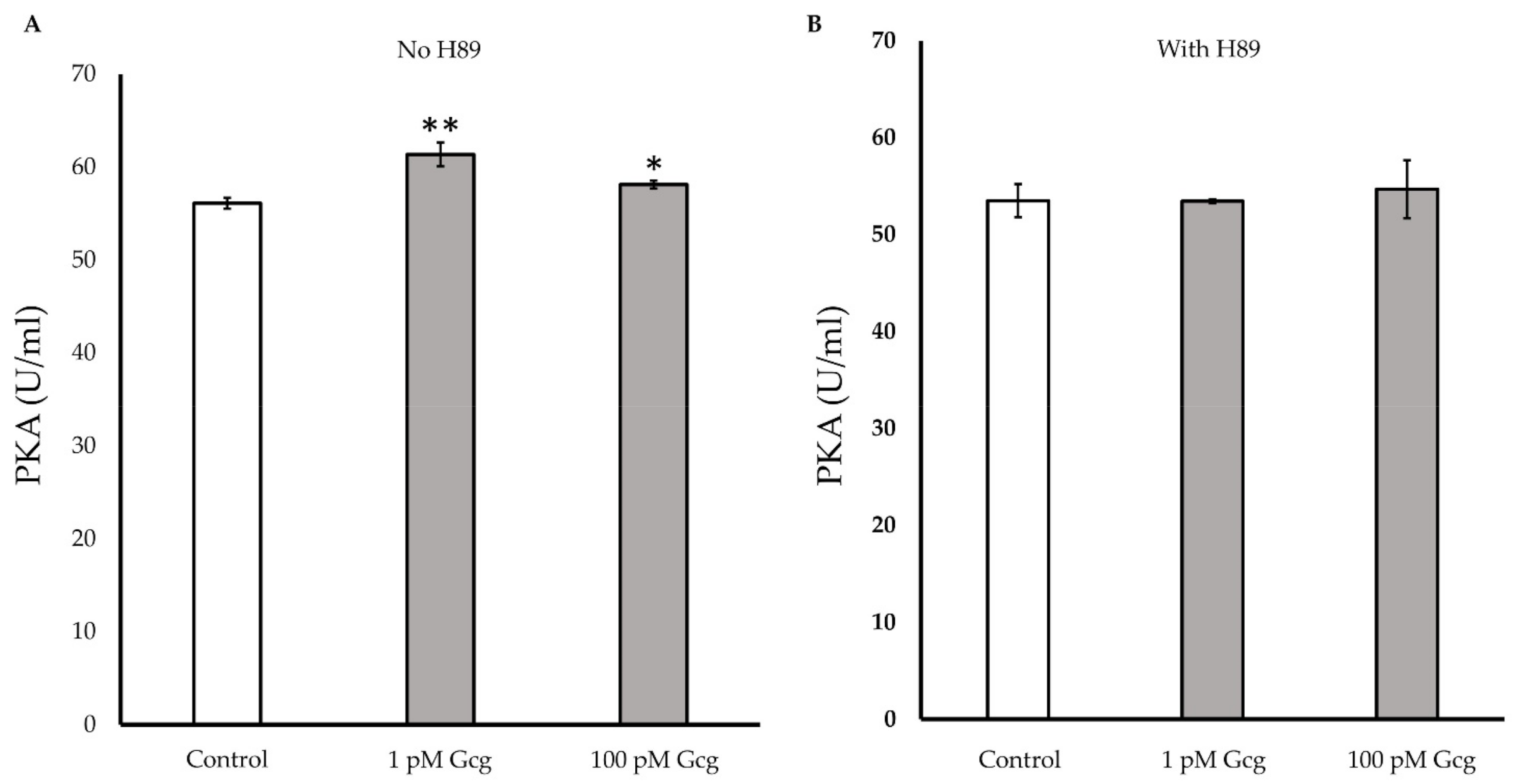
3.7. PKA Activity Was Increased by Glucagon
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Diabetes Control and Complications Trial Research Group. The effect of intensive diabetes therapy on measures of autonomic nervous system function in the Diabetes Control and Complications Trial (DCCT). Diabetologia 1998, 41, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Diabetes Control and Complications Trial Research Group. The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group. Ann. Intern. Med. 1995, 122, 561–568. [Google Scholar] [CrossRef]
- Hotta, N.; Akanuma, Y.; Kawamori, R.; Matsuoka, K.; Oka, Y.; Shichiri, M.; Toyota, T.; Nakashima, M.; Yoshimura, I.; Sakamoto, N.; et al. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: The 3-year, multicenter, comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes Care 2006, 29, 1538–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, C.D.; Alviar, M.J.; Dans, A.L.; Bautista-Velez, G.G.; Villaruz-Sulit, M.V.; Tan, J.J.; Co, H.U.; Bautista, M.R.; Roxas, A.A. Vitamin B for treating peripheral neuropathy. Cochrane Database Syst. Rev. 2008, 16, CD004573. [Google Scholar] [CrossRef] [PubMed]
- Stracke, H.; Gaus, W.; Achenbach, U.; Federlin, K.; Bretzel, R.G. Benfotiamine in diabetic polyneuropathy (BENDIP): Results of a randomised, double blind, placebo-controlled clinical study. Exp. Clin. Endocrinol. Diabetes 2008, 116, 600–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Zhang, B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E671–E678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmley, W.; Glick, G.; Sonnenblick, E. Cardiovascular effects of glucagon in man. N. Engl. J. Med. 1968, 279, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Childress, R.; Chip, J.; Border, J. Hemodynamic effects of glucagon in patients with heart disease. Circulation 1969, 39, 38–47. [Google Scholar]
- Habegger, K.M.; Stemmer, K.; Cheng, C.; Müller, T.D.; Heppner, K.M.; Ottaway, N.; Holland, J.; Hembree, J.L.; Smiley, D.; Gelfanov, V.; et al. Fibroblast Growth Factor 21 Mediates Specific Glucagon Actions. Diabetes 2013, 62, 1453–1463. [Google Scholar] [CrossRef] [Green Version]
- Fanne, R.; Nassar, T.; Mazuz, A.; Waked, O.; HeymanS, N.; Hijazi, N.; Goelman, G.; Higazi, A. Neuroprotection by glucagon: Role of gluconeogenesis. J. Neurosurg. 2011, 114, 85–91. [Google Scholar] [CrossRef]
- Li, Y.; Glotfelty, E.; Namdar, I.; Tweedie, D.; Olson, L.; Hoffer, B.; DiMarchi, R.; Pick, C.; NH, G. Neurotrophic and neuroprotective effects of a monomeric GLP-1/GIP/Gcg receptor triagonist in cellular and rodent models of mild traumatic brain injury. Exp. Neurol. 2020, 324, 113113. [Google Scholar] [CrossRef] [PubMed]
- Motegi, M.; Himeno, T.; Nakai-Shimoda, H.; Inoue, R.; Ozeki, N.; Hayashi, Y.; Sasajima, S.; Mohiuddin, M.; Asano-Hayami, E.; Kato, M.; et al. Deficiency of glucagon gene-derived peptides induces peripheral polyneuropathy in mice. Biochem. Biophys. Res. Commun. 2020, 532, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, M.; Himeno, T.; Inoue, R.; Miura-Yura, E.; Yamada, Y.; Nakai-Shimoda, H.; Asano, S.; Kato, M.; Motegi, M.; Kondo, M.; et al. Glucagon-like peptide-1 receptor agonist protects dorsal root ganglion neurons against oxidative insult. J. Diabetes Res. 2019, 2019, 9426014. [Google Scholar] [CrossRef] [Green Version]
- Himeno, T.; Kamiya, H.; Naruse, K.; Harada, N.; Ozaki, N.; Seino, Y.; Shibata, T.; Kondo, M.; Kato, J.; Okawa, T.; et al. Beneficial effects of exendin-4 on experimental polyneuropathy in diabetic mice. Diabetes 2011, 60, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Goto, Y.; Hotta, N.; Shigeta, Y.; Sakamoto, N.; Kikkawa, R. Effects of an aldose reductase inhibitor, epalrestat, on diabetic neuropathy. Clinical benefit and indication for the drug assessed from the results of a placebo-controlled double-blind study. Biomed. Pharmacother. 1995, 49, 269–277. [Google Scholar] [CrossRef]
- Ziegler, D.; Papanas, N.; Schnell, O.; Nguyen, B.; Nguyen, K.; Kulkantrakorn, K.; Deerochanawong, C. Current concepts in the management of diabetic polyneuropathy. Diabetes Investig. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S.; Yamagishi, S.; Wada, R. Pathology and pathogenetic mechanisms of diabetic neuropathy: Correlation with clinical signs and symptoms. Diabetes Res. Clin. Pract. 2007, 77, S184–S189. [Google Scholar] [CrossRef]
- Vincent, A.M.; McLean, L.L.; Backus, C.; Feldman, E.L. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. Faseb. J. 2005, 19, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.C. Mitochondrial dysfunction and oxidative stress in seizure-induced neuronal cell death. Acta. Neurol. Taiwan 2010, 19, 3–15. [Google Scholar] [PubMed]
- Alam, M.A.; Chowdhury, M.R.; Jain, P.; Sagor, M.A.; Reza, H.M. DPP-4 inhibitor sitagliptin prevents inflammation and oxidative stress of heart and kidney in two kidney and one clip (2K1C) rats. Diabetol. Metab. Syndr. 2015, 7, 107. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Cacicedo, J.; Ido, Y. Impaired nicotinamide adenine dinucleotide (NAD+ ) metabolism in diabetes and diabetic tissues: Implications for nicotinamide-related compound treatment. J. Diabetes Investig. 2020, 11, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Younger, D.; Rosoklija, G.; Hays, A.; Trojaborg, W.; Latov, N. Diabetic peripheral neuropathy: A clinicopathologic and immunohistochemical analysis of sural nerve biopsies. Muscle Nerve 1996, 19, 722–727. [Google Scholar] [CrossRef]
- Conti, G.; Scarpini, E.; Baron, P.; Livraghi, S.; Tiriticco, M.; Bianchi, R.; Vedeler, C.; Scarlato, G. Macrophage infiltration and death in the nerve during the early phases of experimental diabetic neuropathy: A process concomitant with endoneurial induction of IL-1beta and p75NTR. J. Neurol. Sci. 2002, 195, 35–40. [Google Scholar] [CrossRef]
- Leinninger, G.M.; Vincent, A.M.; Feldman, E.L. The role of growth factors in diabetic peripheral neuropathy. J. Peripher. Nerv. Syst. 2004, 9, 26–53. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Tamai, M.; Mori, N. Brain-derived neurotrophic factor prevents axotomized retinal ganglion cell death through MAPK and PI3K signaling pathways. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3319–3326. [Google Scholar] [PubMed]
- Munson, J.B.; Shelton, D.L.; McMahon, S.B. Adult mammalian sensory and motor neurons: Roles of endogenous neurotrophins and rescue by exogenous neurotrophins after axotomy. J. Neurosci. 1997, 17, 470–476. [Google Scholar] [CrossRef]
- Lee, H.; Seo, I.; Suh, D.; Lee, H.; Park, H.T. A novel mechanism of methylglyoxal cytotoxicity in neuroglial cells. J. Neurochem. 2009, 108, 273–284. [Google Scholar] [CrossRef]
- Lee, J.H.; Parveen, A.; Do, M.H.; Kang, M.C.; Yumnam, S.; Kim, S.Y. Molecular mechanisms of methylglyoxal-induced aortic endothelial dysfunction in human vascular endothelial cells. Cell Death. Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Howell, S.K.; O’Dell, R.M.; Wood, M.E.; Touchette, A.D.; Szwergold, B.S. α-Dicarbonyls Increase in the Postprandial Period and Reflect the Degree of Hyperglycemia. Diabetes Care 2001, 24, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.; Huang, D.; Huang, Y.; Hsu, S.; Kang, L.; Shen, C.; Lin, W. Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction. J. Cell Mol. Med. 2016, 20, 1749–1760. [Google Scholar]
- Figarola, J.L.; Singhal, J.; Rahbar, S.; Awasthi, S.; Singhal, S.S. LR-90 prevents methylglyoxal-induced oxidative stress and apoptosis in human endothelial cells. Apoptosis 2014, 19, 776–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radu, B.; Dumitrescu, D.; Mustaciosu, C.; Radu, M. Dual effect of methylglyoxal on the intracellular Ca2+ signaling and neurite outgrowth in mouse sensory neurons. Cell Mol. Neurobiol. 2012, 32, 1047–1057. [Google Scholar] [PubMed]
- McLellan, A.; Thornalley, P.; Benn, J.; Sonksen, P. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. 1994, 87, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapolla, A.; Flamini, R.; Lupo, A.; Aricò, N.; Rugiu, C.; Reitano, R.; Tubaro, M.; Ragazzi, E.; Seraglia, R.; Traldi, P. Evaluation of glyoxal and methylglyoxal levels in uremic patients under peritoneal dialysis. Ann. N. Y. Acad. Sci. 2005, 1043, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Flamini, R.; Dalla Vedova, A.; Senesi, A.; Reitano, R.; Fedele, D.; Basso, E.; Seraglia, R.; Traldi, P. Glyoxal and methylglyoxal levels in diabetic patients: Quantitative determination by a new GC/MS method. Clin. Chem. Lab. Med. 2003, 41, 1166–1173. [Google Scholar] [CrossRef]
- Andersen, S.; Witte, D.; Dalsgaard, E.; Andersen, H.; Nawroth, P.; Fleming, T.; Jensen, T.; Finnerup, N.; Jensen, T.; Lauritzen, T.; et al. Risk factors for incident diabetic polyneuropathy in a cohort with screen-detected type 2 diabetes followed for 13 Years: ADDITION-Denmark. Diabetes Care 2018, 41, 1068–1075. [Google Scholar] [CrossRef] [Green Version]
- Thornalley, P.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef]
- Skapare, E.; Konrade, I.; Liepinsh, E.; Strele, I.; Makrecka, M.; Bierhaus, A.; Lejnieks, A.; Pirags, V.; Dambrova, M. Association of reduced glyoxalase 1 activity and painful peripheral diabetic neuropathy in type 1 and 2 diabetes mellitus patients. J. Diabetes Complicat. 2013, 27, 262–267. [Google Scholar] [CrossRef]
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.; Lachin, J.; Monnier, V. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes 2015, 64, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Mi, R.; Haughey, N.; Oz, M.; Höke, A. Immortalization and characterization of a nociceptive dorsal root ganglion sensory neuronal line. J. Peripher. Nerv. Syst. 2007, 12, 121–130. [Google Scholar]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-based flow cytometry for detecting mitochondrial ROS. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radio, N.; Breier, J.; Shafer, T.; Mundy, W. Assessment of chemical effects on neurite outgrowth in PC12 cells using high content screening. Toxicol. Sci. 2008, 105, 106–118. [Google Scholar] [PubMed] [Green Version]
- Pradelles, P.; Grassi, J.; Chabardes, D.; Guiso, N. Enzyme immunoassays of adenosine cyclic 3′,5′-monophosphate and guanosine cyclic 3′,5′-monophosphate using acetylcholinesterase. Anal. Chem. 1989, 61, 447–453. [Google Scholar] [CrossRef]
- Iwai, T.; Ito, S.; Tanimitsu, K.; Udagawa, S.; Oka, J. Glucagon-like peptide-1 inhibits LPS-induced IL-1beta production in cultured rat astrocytes. Neurosci. Res. 2006, 55, 352–360. [Google Scholar] [CrossRef]
- Koschinski, A.; Zaccolo, M. Activation of PKA in cell requires higher concentration of cAMP than in vitro: Implications for compartmentalization of cAMP signalling. Sci. Rep. 2017, 7, 1–12. [Google Scholar]
- Limbutara, K.; Kelleher, A.; Yang, C.; Raghuram, V.; Knepper, M. Phosphorylation changes in response to kinase inhibitor H89 in PKA-null cells. Sci. Rep. 2019, 9, 2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, E.; Yusta, B.; Streutker, C.; Baggio, L.; Koehler, J.; Charron, M.; Drucker, D. Glucagon receptor signaling is essential for control of murine hepatocyte survival. Gastroenterology 2008, 135, 2096–2106. [Google Scholar] [CrossRef] [Green Version]
- Jelinek, L.; Lok, S.; Rosenberg, G.; Smith, R.; Grant, F.; Biggs, S.; Bensch, P.; Kuijper, J.; Sheppard, P.; Sprecher, C. Expression cloning and signaling properties of the rat glucagon receptor. Science 1993, 259, 1614–1616. [Google Scholar] [CrossRef]
- Waltereit, R.; Weller, M. Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol. Neurobiol. 2003, 27, 99–106. [Google Scholar]
- Savchenko, A.; Barnes, S.; Kramer, R.H. Cyclic-nucleotide-gated channels mediate synaptic feedback by nitric oxide. Nature 2020, 390, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, M.S.; Kim, K.-Y.; Bu, J.H.; Nam, H.S.; Jeong, S.W.; Park, T.L.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.-K. Elevated intracellular cAMP exacerbates vulnerability to oxidative stress in optic nerve head astrocytes. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Cai, D.; Dai, H.; McAtee, M.; Hoffman, P.; Bregman, B.; Filbin, M. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 2002, 34, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Kedia, N. Treatment of severe diabetic hypoglycemia with glucagon: An underutilized therapeutic approach. Diabetes Metab. Syndr. Obes. 2011, 4, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohiuddin, M.S.; Himeno, T.; Yamada, Y.; Morishita, Y.; Kondo, M.; Tsunekawa, S.; Kato, Y.; Nakamura, J.; Kamiya, H. Glucagon Prevents Cytotoxicity Induced by Methylglyoxal in a Rat Neuronal Cell Line Model. Biomolecules 2021, 11, 287. https://doi.org/10.3390/biom11020287
Mohiuddin MS, Himeno T, Yamada Y, Morishita Y, Kondo M, Tsunekawa S, Kato Y, Nakamura J, Kamiya H. Glucagon Prevents Cytotoxicity Induced by Methylglyoxal in a Rat Neuronal Cell Line Model. Biomolecules. 2021; 11(2):287. https://doi.org/10.3390/biom11020287
Chicago/Turabian StyleMohiuddin, Mohammad Sarif, Tatsuhito Himeno, Yuichiro Yamada, Yoshiaki Morishita, Masaki Kondo, Shin Tsunekawa, Yoshiro Kato, Jiro Nakamura, and Hideki Kamiya. 2021. "Glucagon Prevents Cytotoxicity Induced by Methylglyoxal in a Rat Neuronal Cell Line Model" Biomolecules 11, no. 2: 287. https://doi.org/10.3390/biom11020287
APA StyleMohiuddin, M. S., Himeno, T., Yamada, Y., Morishita, Y., Kondo, M., Tsunekawa, S., Kato, Y., Nakamura, J., & Kamiya, H. (2021). Glucagon Prevents Cytotoxicity Induced by Methylglyoxal in a Rat Neuronal Cell Line Model. Biomolecules, 11(2), 287. https://doi.org/10.3390/biom11020287