
Open-Bundle Structure as the Unfolding Intermediate of Cytochrome c′ Revealed by Small Angle Neutron Scattering

 ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Preparation for the SANS Experiments
2.2. SANS Measurements
2.3. SANS Data Reduction and Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Joint-Clubs Model
Appendix B. Molecular Weight Estimation
Appendix C. Ab Initio Bead Modeling and Analysis of the Bead Model
Appendix D. Estimation of the Aggregated Particle Fraction at pD ~13
References
- Anfinsen, C.B.; Haber, E.; Sela, M.; White, F.H. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. USA 1961, 47, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ohgushi, M.; Wada, A. “Molten-globule state”: A compact form of globular proteins with mobile side-chains. FEBS Lett. 1983, 164, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, M.; Nishii, I.; Fujisawa, T.; Ueki, T.; Tokunaga, F.; Goto, Y. Structural characterization of the molten globule and native states of apomyoglobin by solution x-ray scattering. J. Mol. Biol. 1995, 249, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Bhuyan, A.K. Off-pathway status for the alkali molten globule of horse ferricytochrome c. Biochemistry 2010, 49, 7764–7773. [Google Scholar] [CrossRef]
- Arai, M.; Kuwajima, K. Role of the molten globule state in protein folding. Adv. Protein Chem. 2000, 53, 209–282. [Google Scholar]
- Hamada, D.; Hoshino, M.; Kataoka, M.; Goto, Y.; Fink, A.L. Intermediate Conformational States of Apocytochrome c. Biochemistry 1993, 32, 10351–10358. [Google Scholar] [CrossRef]
- Kiefhaber, T. Kinetic traps in lysozyme folding. Proc. Natl. Acad. Sci. USA 1995, 92, 9029–9033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elöve, G.A.; Bhuya, A.K.; Roder, H. Kinetic Mechanism of Cytochrome c Folding: Involvement of the Heme and Its Ligands. Biochemistry 1994, 33, 6925–6935. [Google Scholar] [CrossRef]
- Kamatari, Y.O.; Konno, T.; Kataoka, M.; Akasaka, K. The methanol-induced transition and the expanded helical conformation in hen lysozyme. Protein Sci. 1998, 7, 681–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamill, A.C.; Wang, S.C.; Lee, C.T. Probing lysozyme conformation with light reveals a new folding intermediate. Biochemistry 2005, 44, 15139–15149. [Google Scholar] [CrossRef]
- Jamin, M.; Baldwin, R.L. Refolding and unfolding kinetics of the equilibrium folding intermediate of apomyoglobin. Nat. Struct. Biol. 1996, 3, 613–618. [Google Scholar] [CrossRef]
- Kitahara, R.; Akasaka, K. Close identity of a pressure-stabilized intermediate with a kinetic intermediate in protein folding. Proc. Natl. Acad. Sci. USA 2003, 100, 3167–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Hagihara, Y.; Hamada, D.; Hoshino, M.; Nishii, I. Acid-Induced Unfolding and Refolding Transitions of Cytochrome c: A Three-State Mechanism in H2O and D2O. Biochemistry 1993, 32, 11878–11885. [Google Scholar] [CrossRef]
- Ikeguchi, M.; Kuwajima, K.; Mitani, M.; Sugai, S. Evidence for Identity between the Equilibrium Unfolding Intermediate and a Transient Folding Intermediate: A Comparative Study of the Folding Reactions of α-Lactalbumin and Lysozyme. Biochemistry 1986, 25, 6965–6972. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hodgson, K.O.; Doniach, S. A lysozyme folding intermediate revealed by solution X-ray scattering. J. Mol. Biol. 1996, 261, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.M.G.; Maity, H.; Rumbley, J.N.; Lin, Y.; Englander, S.W. Order of Steps in the Cytochrome c Folding Pathway: Evidence for a Sequential Stabilization Mechanism. J. Mol. Biol. 2006, 359, 1410–1419. [Google Scholar] [CrossRef]
- Hu, W.; Walters, B.T.; Kan, Z.Y.; Mayne, L.; Rosen, L.E.; Marqusee, S.; Englander, S.W. Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. USA 2013, 110, 7684–7689. [Google Scholar] [CrossRef] [Green Version]
- Hoang, L.; Bedard, S.; Krishna, M.M.G.; Lin, Y.; Englander, S.W. Cytochrome c folding pathway: Kinetic native-state hydrogen exchange. Proc. Natl. Acad. Sci. USA 2002, 99, 12173–12178. [Google Scholar] [CrossRef] [Green Version]
- Freddolino, P.L.; Harrison, C.B.; Liu, Y.; Schulten, K. Challenges in protein-folding simulations. Nat. Phys. 2010, 6, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilquin, B.; Guilbert, C.; Perahia, D. Unfolding of hen egg lysozyme by molecular dynamics simulations at 300K: Insight into the role of the interdomain interface. Proteins Struct. Funct. Genet. 2000, 41, 58–74. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How fast-folding proteins fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I.; Koch, M.H.J. Advances in structure analysis using small-angle scattering in solution. Curr. Opin. Struct. Biol. 2002, 12, 654–660. [Google Scholar] [CrossRef]
- Lattman, E.E. Small angle scattering studies of protein folding. Curr. Opin. Struct. Biol. 1994, 4, 87–92. [Google Scholar] [CrossRef]
- Akiyama, S.; Takahashi, S.; Kimura, T.; Ishimori, K.; Morishima, I.; Nishikawa, Y.; Fujisawa, T. Conformational landscape of cytochrome c folding studied by microsecond-resolved small-angle x-ray scattering. Proc. Natl. Acad. Sci. USA 2002, 99, 1329–1334. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Inoko, Y. Size-exclusion chromatography combined with small-angle X-ray scattering optics. J. Chromatogr. A 2009, 1216, 7461–7465. [Google Scholar] [CrossRef]
- Koutsioubas, A.; Pérez, J. Incorporation of a hydration layer in the ‘dummy atom’ ab initio structural modelling of biological macromolecules. J. Appl. Crystallogr. 2013, 46, 1884–1888. [Google Scholar] [CrossRef]
- Koutsioubas, A.; Jaksch, S.; Pérez, J. DENFERT version 2: Extension of ab initio structural modelling of hydrated biomolecules to the case of small-angle neutron scattering data. J. Appl. Crystallogr. 2016, 49, 690–695. [Google Scholar] [CrossRef]
- Maltempo, M.M.; Moss, T.H. The spin 3/2 state and quantum spin mixtures in haem proteins. Q. Rev. Biophys. 1976, 9, 181–215. [Google Scholar] [CrossRef]
- Maltempo, M.M.; Moss, T.H.; Cusanovich, M.A. Magnetic studies on the changes in the iron environment in Chromatium ferricytochrome c′. BBA-Protein Struct. 1974, 342, 290–305. [Google Scholar] [CrossRef]
- Yoshimura, T.; Suzuki, S.; Nakahara, A.; Iwasaki, H.; Masuko, M.; Matsubara, T. Spectral Properties of Nitric Oxide Complexes of Cytochrome c’ from Alcaligenes sp. NCIB 11015. Biochemistry 1986, 25, 2436–2442. [Google Scholar] [CrossRef]
- Weiss, R.; Gold, A.; Terner, J. Cytochromes c′: Biological models for the S = 3/2,5/2 spin-state admixture? Chem. Rev. 2006, 106, 2550–2579. [Google Scholar] [CrossRef]
- Takashina, A.; Tiedemann, M.T.; Unno, M.; Yamaguchi, T.; Stillman, M.J.; Kohzuma, T. The pH Dependent Protein Structure Transitions and Related Spin-State Transition of Cytochrome c′ from Alcaligenes xylosoxidans NCIMB 11015. Bull. Chem. Soc. Jpn. 2017, 90, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Covington, A.K.; Paabo, M.; Robinson, R.A.; Bates, R.G. Use of the Glass Electrode in Deuterium Oxide and the Relation between the Standardized pD (paD) Scale and the Operational pH in Heavy Water. Anal. Chem. 1968, 40, 700–706. [Google Scholar] [CrossRef]
- Feoktystov, A.V.; Frielinghaus, H.; Di, Z.; Jaksch, S.; Pipich, V.; Appavou, M.-S.; Babcock, E.; Hanslik, R.; Engels, R.; Kemmerling, G.; et al. KWS-1 high-resolution small-angle neutron scattering instrument at JCNS: Current state. J. Appl. Crystallogr. 2015, 48, 61–70. [Google Scholar] [CrossRef]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Debye, P. Molecular-Weight Determination by Light Scattering. J. Phys. Colloid Chem. 1946, 51, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Mortensen, K.; Talmon, Y. Cryo-TEM and SANS Microstructural Study of Pluronic Polymer Solutions. Macromolecules 1995, 28, 8829–8834. [Google Scholar] [CrossRef]
- Beaucage, G. Small-Angle Scattering from Polymeric Mass Fractals of Arbitrary Mass-Fractal Dimension. J. Appl. Crystallogr. 1996, 29, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Dobbs, A.J.; Anderson, B.F.; Faber, H.R.; Baker, E.N. Three-dimensional structure of cytochrome c′ from two Alcaligenes species and the implications for four-helix bundle structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 356–368. [Google Scholar] [CrossRef]
- Svergun, D.I.; Barberato, C.; Koch, M.H.J.; Fetler, L.; Vachette, P. Large differences are observed between the crystal and solution quaternary structures of allosteric aspartate transcarbamylase in the R state. Proteins 1997, 27, 110–117. [Google Scholar] [CrossRef]
- Fetler, L.; Vachette, P. The allosteric activator Mg-ATP modifies the quaternary structure of the R-state of Escherichia coli aspartate transcarbamylase without altering the T ⟷ R equilibrium. J. Mol. Biol. 2001, 309, 817–832. [Google Scholar] [CrossRef]
- Meisburger, S.P.; Thomas, W.C.; Watkins, M.B.; Ando, N. X-ray Scattering Studies of Protein Structural Dynamics. Chem. Rev. 2017, 117, 7615–7672. [Google Scholar] [CrossRef]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doniach, S. Changes in biomolecular conformation seen by small angle X-ray scattering. Chem. Rev. 2001, 101, 1763–1778. [Google Scholar] [CrossRef] [PubMed]
- Burger, V.M.; Arenas, D.J.; Stultz, C.M. A Structure-free Method for Quantifying Conformational Flexibility in proteins. Sci. Rep. 2016, 6, 29040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonas, E.; Svergun, D.I. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 2007, 40, s245–s249. [Google Scholar] [CrossRef] [Green Version]
- Hermans, J.; Hermans, J.J. Light Scattering by Zig-zag and Worm-like Chains. J. Phys. Chem. 1958, 62, 1543–1546. [Google Scholar] [CrossRef]
- Lages, S.; Goerigk, G.; Huber, K. SAXS and ASAXS on Dilute Sodium Polyacrylate Chains Decorated with Lead Ions. Macromolecules 2013, 46, 3570–3580. [Google Scholar] [CrossRef]
- Wriggers, W. Conventions and workflows for using Situs. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, S.; Hattori, Y.; Nagao, S.; Taketa, M.; Komori, H.; Kamikubo, H.; Wang, Z.; Takahashi, I.; Negi, S.; Sugiura, Y.; et al. Cytochrome c polymerization by successive domain swapping at the C-terminal helix. Proc. Natl. Acad. Sci. USA 2010, 107, 12854–12859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margoliash, E.; Lustgarten, J. Interconversion of Horse Heart Cytochrome c Monomer and Polymers. J. Biol. Chem. 1962, 237, 3397–3405. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Svergun, D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005, 89, 1237–1250. [Google Scholar] [CrossRef] [Green Version]
- Takashina, A.; Unno, M.; Kohzuma, T. X-ray Crystallographic Elucidation for the Alkaline High-spin State Transition of Iron (III) Cytochrome c′ from Alcaligenes xylosoxidans NCIMB 11015. Chem. Lett. 2015, 44, 268–270. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Lynn, G.W.; Guo, J.; Melnichenko, Y.B.; Wignall, G.D.; McClain, J.B.; DeSimone, J.M.; Johnson, C.S. NMR and SANS Studies of Aggregation and Microemulsion Formation by Phosphorus Fluorosurfactants in Liquid and Supercritical Carbon Dioxide. J. Phys. Chem. B 2005, 109, 10261–10269. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, L. Correlations in space and time and born approximation scattering in systems of interacting particles. Phys. Rev. 1954, 95, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Svergun, D.I.; Richard, S.; Koch, M.H.J.; Sayers, Z.; Kuprin, S.; Zaccai, G. Protein hydration in solution: Experimental observation by x-ray and neutron scattering. Proc. Natl. Acad. Sci. USA 1998, 95, 2267–2272. [Google Scholar] [CrossRef] [Green Version]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
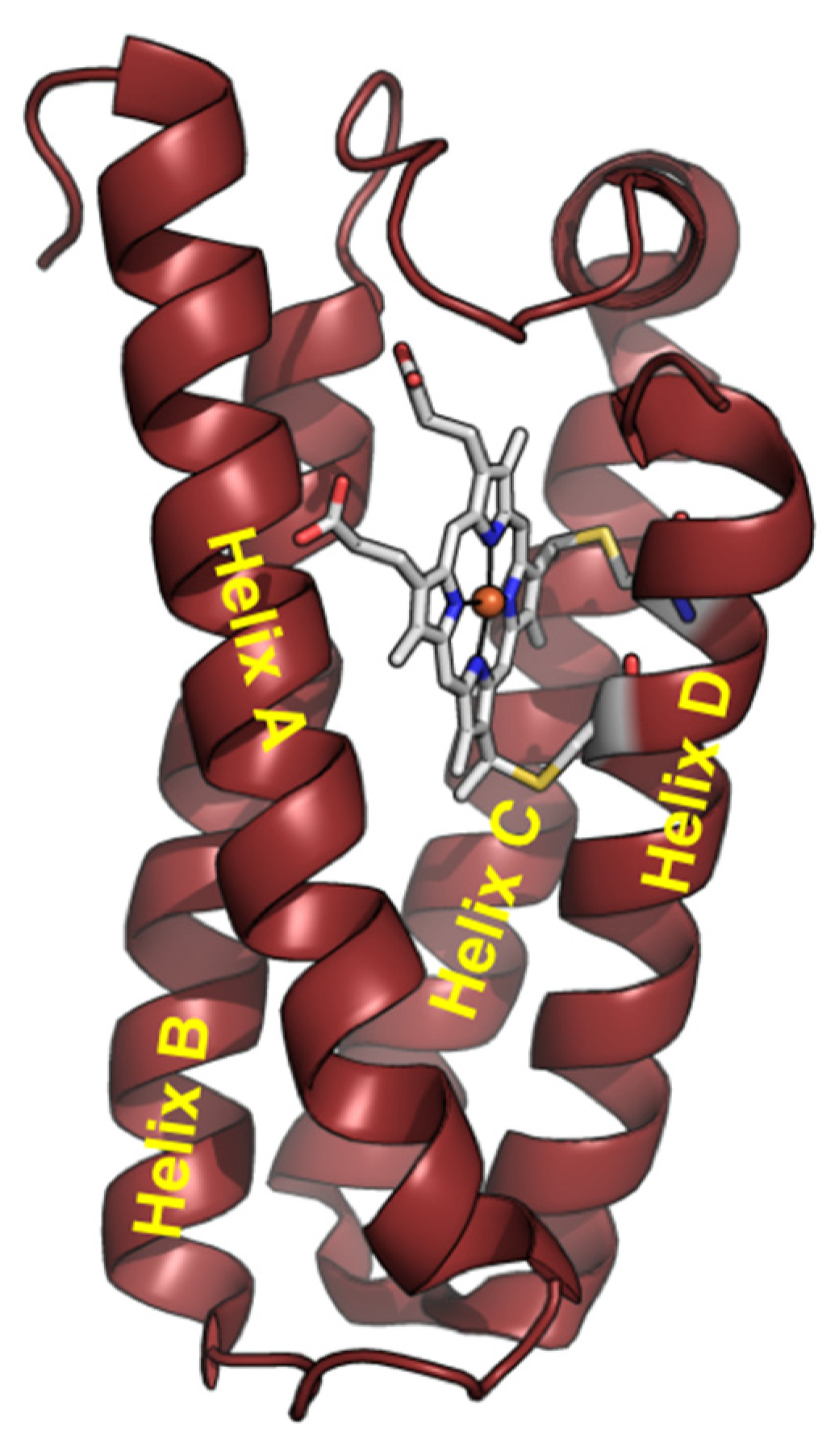{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
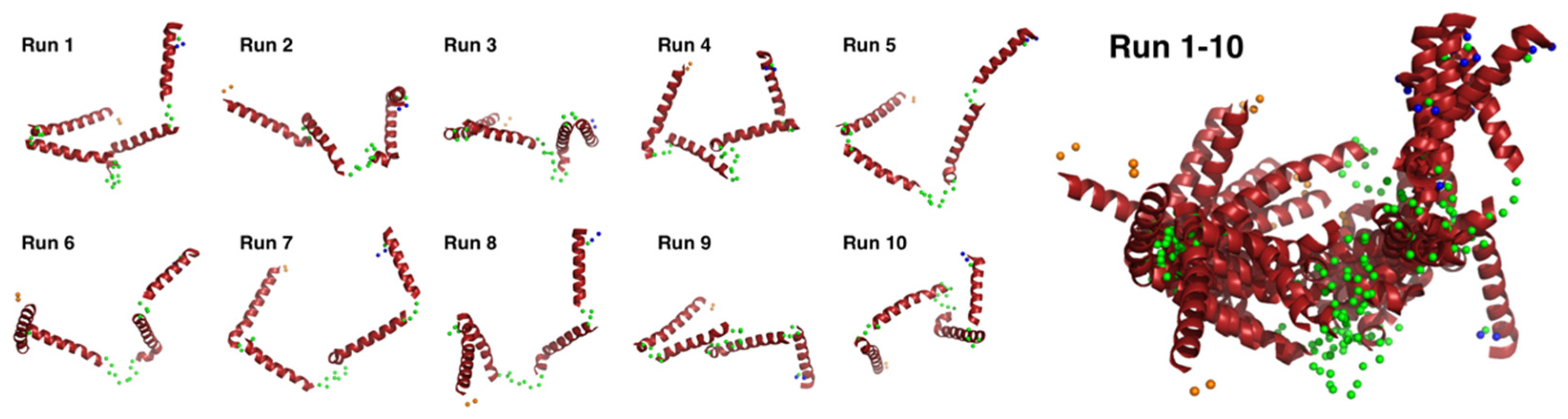{kind=link}
| Conditions | Rg/Å | ||
|---|---|---|---|
| Guinier | P(r) | Debye | |
| pD 1.7 | 23.02 ± 0.57 | 25.03 ± 0.19 | 25.65 ± 0.15 |
| pD 6.4 | 18.93 ± 0.45 | 18.24 ± 0.08 | n/a |
| pD 9.6 | 19.24 ± 0.49 | 18.10 ± 0.05 | n/a |
| pD ~13 | 27.58 ± 0.59 | 25.54 ± 0.60 | n/a |
| Conditions | Rg/Å | ||
|---|---|---|---|
| Guinier | P(r) | ||
| pH 6.0 | Monomer | 14.0 | 15.6 |
| Dimer | 17.5 | 19.1 | |
| pH 10.4 | Monomer | 13.6 | 15.5 |
| Dimer | 17.3 | 19.1 | |
| Title 1 | C (mg/mL) | Guinier | P(r) | ||
|---|---|---|---|---|---|
| I(0) | MW Ratio | I(0) | MW Ratio | ||
| pD 1.7 | 5.5 | 0.078 | 1.0 | 0.080 | 1.0 |
| pD 6.4 | 5.5 | 0.165 | 2.1 | 0.161 | 2.0 |
| pD 9.6 | 5.3 | 0.141 | 1.9 | 0.139 | 1.8 |
| pD ~13 | 5.3 | 0.088 | 1.2 | 0.080 | 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, T.; Akao, K.; Koutsioubas, A.; Frielinghaus, H.; Kohzuma, T. Open-Bundle Structure as the Unfolding Intermediate of Cytochrome c′ Revealed by Small Angle Neutron Scattering. Biomolecules 2022, 12, 95. https://doi.org/10.3390/biom12010095
Yamaguchi T, Akao K, Koutsioubas A, Frielinghaus H, Kohzuma T. Open-Bundle Structure as the Unfolding Intermediate of Cytochrome c′ Revealed by Small Angle Neutron Scattering. Biomolecules. 2022; 12(1):95. https://doi.org/10.3390/biom12010095
Chicago/Turabian StyleYamaguchi, Takahide, Kouhei Akao, Alexandros Koutsioubas, Henrich Frielinghaus, and Takamitsu Kohzuma. 2022. "Open-Bundle Structure as the Unfolding Intermediate of Cytochrome c′ Revealed by Small Angle Neutron Scattering" Biomolecules 12, no. 1: 95. https://doi.org/10.3390/biom12010095
APA StyleYamaguchi, T., Akao, K., Koutsioubas, A., Frielinghaus, H., & Kohzuma, T. (2022). Open-Bundle Structure as the Unfolding Intermediate of Cytochrome c′ Revealed by Small Angle Neutron Scattering. Biomolecules, 12(1), 95. https://doi.org/10.3390/biom12010095