
DOTA: Deep Learning Optimal Transport Approach to Advance Drug Repositioning for Alzheimer’s Disease

Abstract
:1. Introduction
2. Materials and Methods
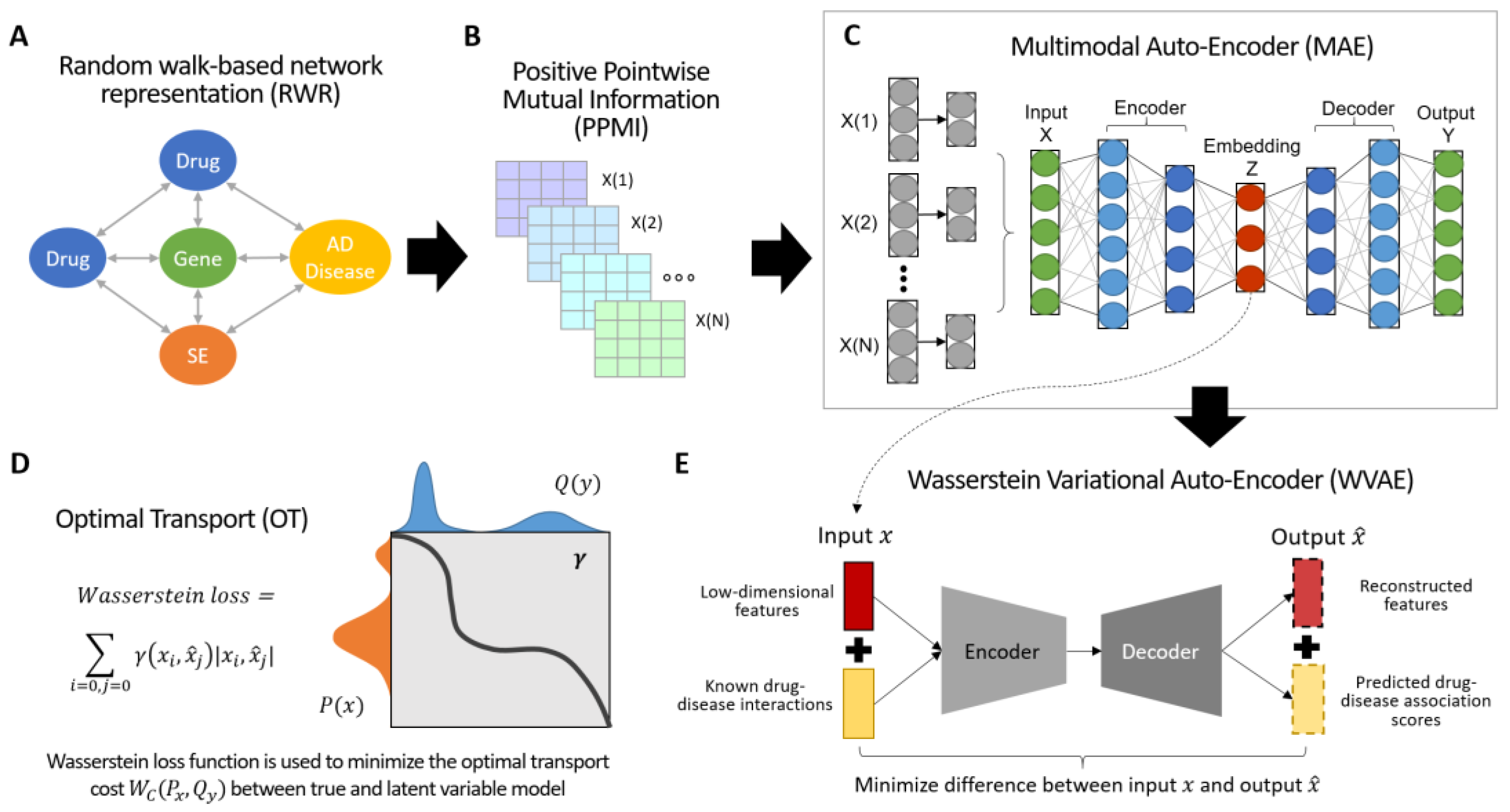
2.1. Assembling Drug–Target–Sideeffects–Disease Networks
2.2. Network Representation and Fusion
2.3. Drug–Disease Predictions with Optimal Trasport
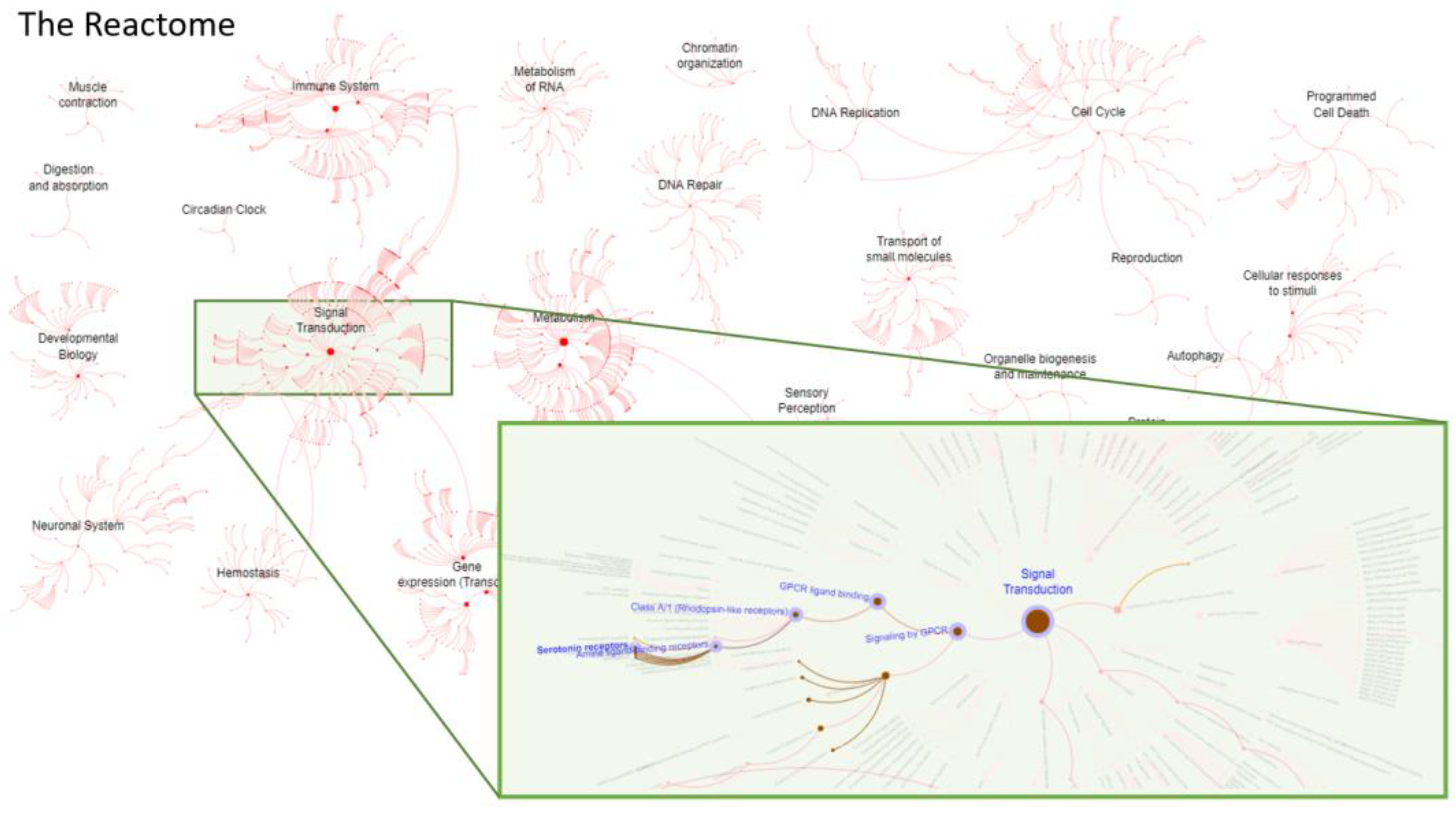
2.4. Analysis of the Human Reactome
2.5. Analysis of the Human Diseasome
3. Results
3.1. Overview of DOTA
3.2. Constructing and Integrating Drug Networks
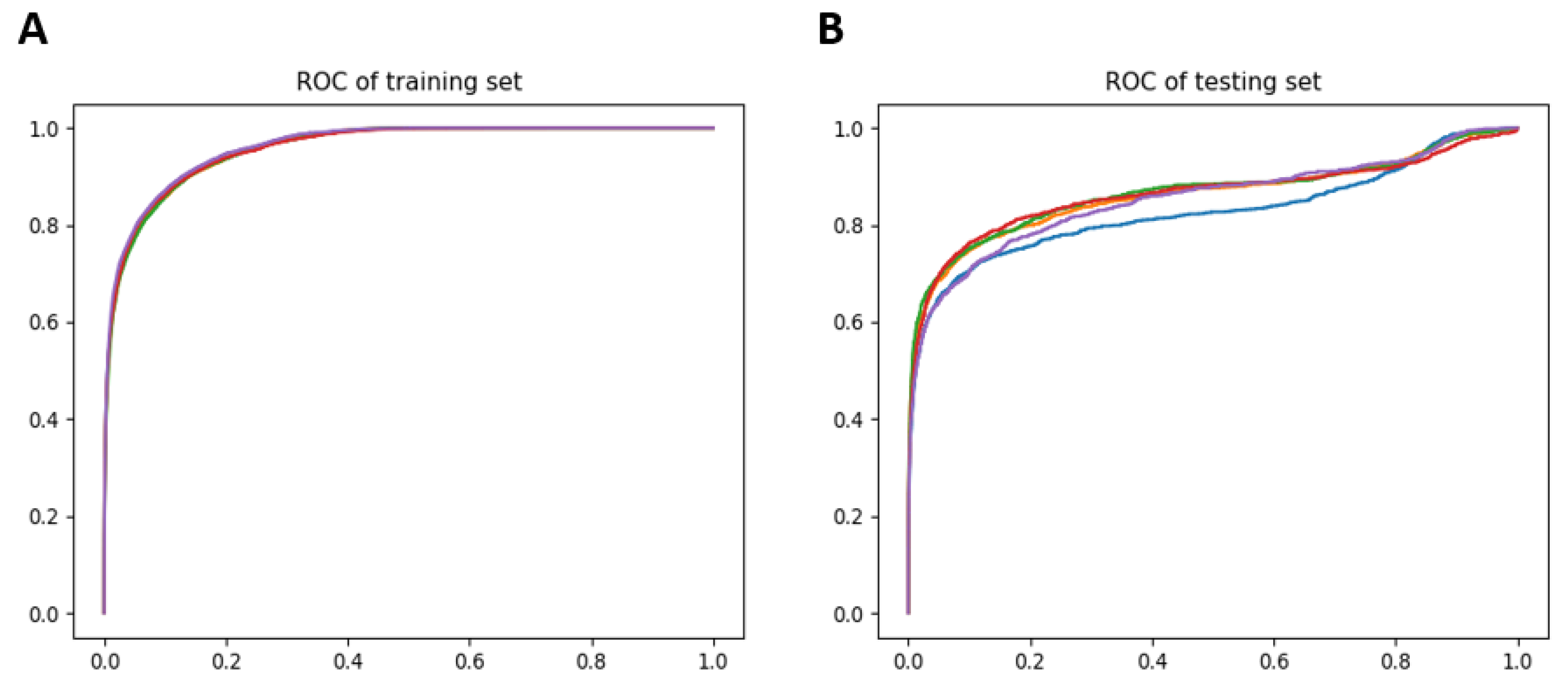
3.3. Drug Predictions and Association Using Optimal Transport
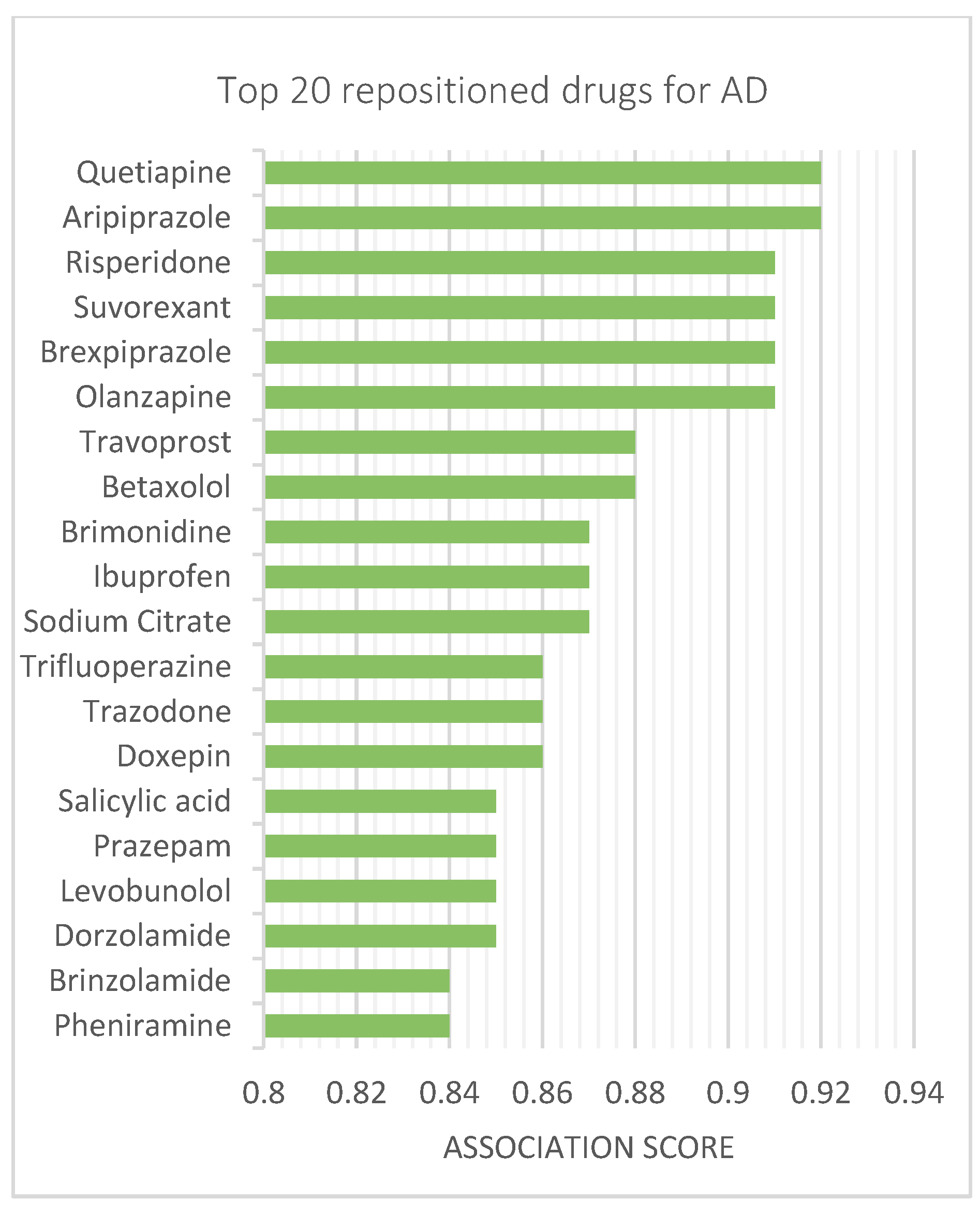
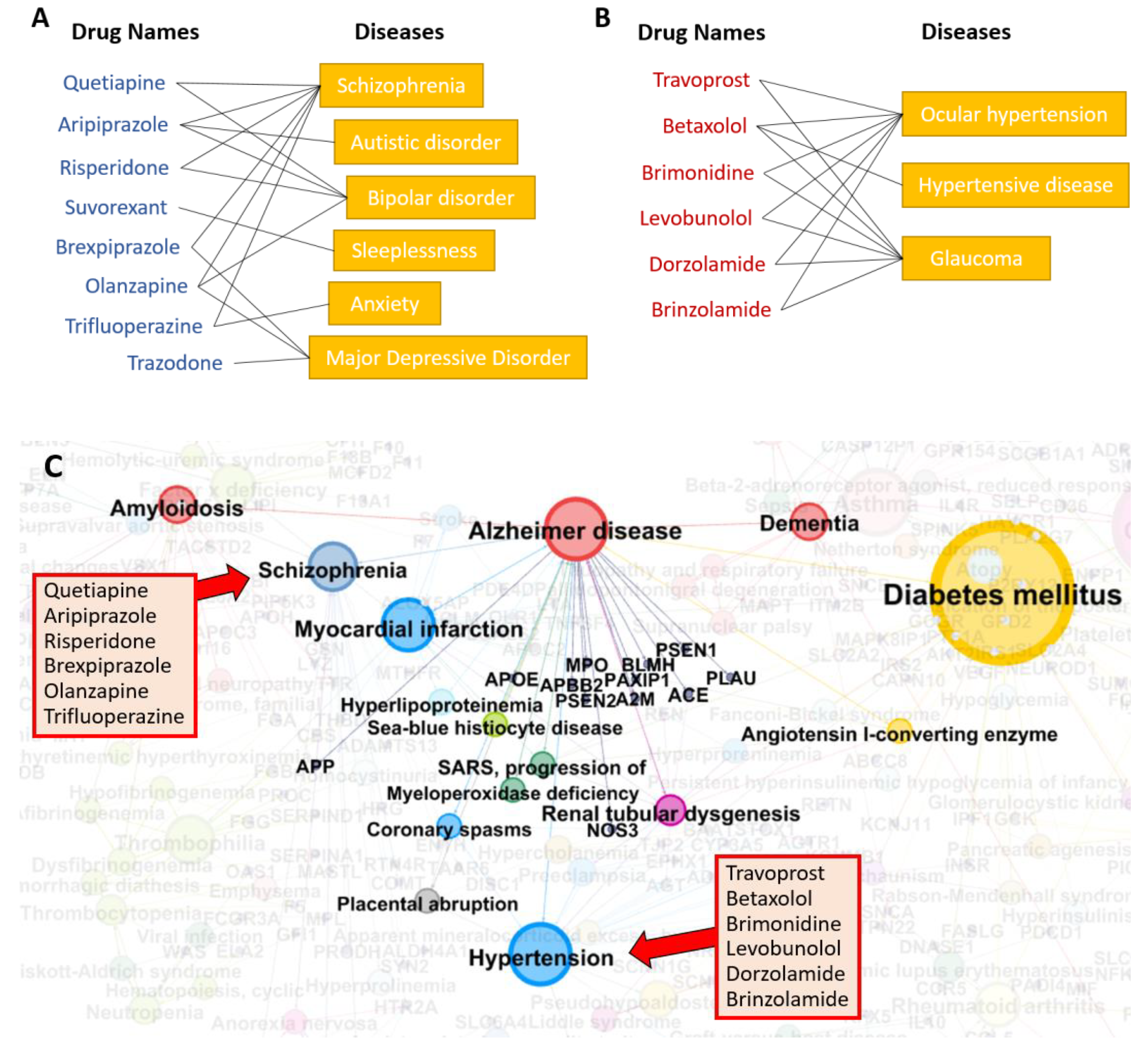
3.4. Repositionig Results and Validation
3.5. Reactome Analysis—Functional and Biological Targets of Repositioned Drugs
3.6. Quantifying Anticholinergic Burden and Sedative Load of Repositioned Drugs
3.7. Diseasome Analysis—Relationships between AD and Other Diseases
3.8. Clinical Analysis of Candidate AD Drugs and Their Effects on Circadian Patterns
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ziegler-Graham, K.; Brookmeyer, R.; Johnson, E.; Arrighi, H.M. Worldwide variation in the doubling time of Alzheimer’s disease incidence rates. Alzheimers Dement. 2008, 4, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Olivares, D.; Deshpande, V.K.; Shi, Y.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T.; Huang, X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr. Alzheimer Res. 2012, 9, 746–758. [Google Scholar] [CrossRef]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sanchez-Lopez, E.; Garcia, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plast. 2016, 2016, 8501693. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef]
- Casey, D.A.; Antimisiaris, D.; O’Brien, J. Drugs for Alzheimer’s disease: Are they effective? Pharm. Ther. 2010, 35, 208–211. [Google Scholar]
- Zhang, M.; Schmitt-Ulms, G.; Sato, C.; Xi, Z.; Zhang, Y.; Zhou, Y.; St George-Hyslop, P.; Rogaeva, E. Drug Repositioning for Alzheimer’s Disease Based on Systematic ‘omics’ Data Mining. PLoS ONE 2016, 11, e0168812. [Google Scholar] [CrossRef] [Green Version]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, G.K.; Black, S.E.; Hendrix, S.B.; Zavitz, K.H.; Swabb, E.A.; Laughlin, M.A.; Tarenflurbil Phase II Study Investigators. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: A randomised phase II trial. Lancet Neurol. 2008, 7, 483–493. [Google Scholar] [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H.; Group, T.P.S. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Hedskog, L.; Petersen, C.A.H.; Winblad, B.; Ankarcrona, M. Dimebon (latrepirdine) enhances mitochondrial function and protects neuronal cells from death. J. Alzheimer’s Dis. 2010, 21, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Onofrio, G.; Panza, F.; Frisardi, V.; Solfrizzi, V.; Imbimbo, B.P.; Paroni, G.; Cascavilla, L.; Seripa, D.; Pilotto, A. Advances in the identification of γ-secretase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Discov. 2012, 7, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.; Williams, G.; Ballard, C. Drug repositioning in Alzheimer’s disease. Front. Biosci. 2015, 7, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andronis, C.; Sharma, A.; Virvilis, V.; Deftereos, S.; Persidis, A. Literature mining, ontologies and information visualization for drug repurposing. Brief Bioinform. 2011, 12, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Chindelevitch, L.; Ziemek, D.; Enayetallah, A.; Randhawa, R.; Sidders, B.; Brockel, C.; Huang, E.S. Causal reasoning on biological networks: Interpreting transcriptional changes. Bioinformatics 2012, 28, 1114–1121. [Google Scholar] [CrossRef]
- Luo, H.; Chen, J.; Shi, L.; Mikailov, M.; Zhu, H.; Wang, K.; He, L.; Yang, L. DRAR-CPI: A server for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome. Nucleic Acids Res. 2011, 39, W492–W498. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Agarwal, P. Systematic drug repositioning based on clinical side-effects. PLoS ONE 2011, 6, e28025. [Google Scholar] [CrossRef]
- Yang, L.; Wang, K.; Chen, J.; Jegga, A.G.; Luo, H.; Shi, L.; Wan, C.; Guo, X.; Qin, S.; He, G.; et al. Exploring off-targets and off-systems for adverse drug reactions via chemical-protein interactome--clozapine-induced agranulocytosis as a case study. PLoS Comput. Biol. 2011, 7, e1002016. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frolich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaques, N.; Taylor, S.; Sano, A.; Picard, R. Multimodal autoencoder: A deep learning approach to filling in missing sensor data and enabling better mood prediction. In Proceedings of the 2017 Seventh International Conference on Affective Computing and Intelligent Interaction (ACII), San Antonio, TX, USA, 23–26 October 2017; pp. 202–208. [Google Scholar]
- Wang, H.; Wang, J.; Dong, C.; Lian, Y.; Liu, D.; Yan, Z. A Novel Approach for Drug-Target Interactions Prediction Based on Multimodal Deep Autoencoder. Front. Pharmacol. 2020, 10, 1592. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xu, X.; Qiu, Y.; Xia, J.; Zhang, W.; Liu, S. A multimodal deep learning framework for predicting drug–drug interaction events. Bioinformatics 2020, 36, 4316–4322. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S. Circadian clock disruption in neurodegenerative diseases: Cause and effect? Front. Pharmacol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazer, D.G.; Hays, J.C.; Foley, D.J. Sleep complaints in older adults: A racial comparison. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, M280–M284. [Google Scholar] [CrossRef]
- Van Someren, E.J. Circadian and sleep disturbances in the elderly. Exp. Gerontol. 2000, 35, 1229–1237. [Google Scholar] [CrossRef]
- Hatfield, C.F.; Herbert, J.; van Someren, E.J.; Hodges, J.R.; Hastings, M.H. Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer’s dementia. Brain 2004, 127, 1061–1074. [Google Scholar] [CrossRef] [Green Version]
- Bedrosian, T.A.; Nelson, R.J. Sundowning syndrome in aging and dementia: Research in mouse models. Exp. Neurol. 2013, 243, 67–73. [Google Scholar] [CrossRef]
- Weldemichael, D.A.; Grossberg, G.T. Circadian rhythm disturbances in patients with Alzheimer’s disease: A review. Int. J. Alzheimer’s Dis. 2010, 2010, 716453. [Google Scholar] [CrossRef] [Green Version]
- Coogan, A.N.; Schutova, B.; Husung, S.; Furczyk, K.; Baune, B.T.; Kropp, P.; Hassler, F.; Thome, J. The circadian system in Alzheimer’s disease: Disturbances, mechanisms, and opportunities. Biol. Psychiatry 2013, 74, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Tranah, G.J.; Blackwell, T.; Stone, K.L.; Ancoli-Israel, S.; Paudel, M.L.; Ensrud, K.E.; Cauley, J.A.; Redline, S.; Hillier, T.A.; Cummings, S.R.; et al. Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann. Neurol. 2011, 70, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, A.S.; Yu, L.; Kowgier, M.; Schneider, J.A.; Buchman, A.S.; Bennett, D.A. Modification of the relationship of the apolipoprotein E epsilon4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 2013, 70, 1544–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Videnovic, A.; Lazar, A.S.; Barker, R.A.; Overeem, S. ‘The clocks that time us’--circadian rhythms in neurodegenerative disorders. Nat. Rev. Neurol. 2014, 10, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 2016, 354, 1004–1008. [Google Scholar] [CrossRef] [Green Version]
- Ambree, O.; Touma, C.; Gortz, N.; Keyvani, K.; Paulus, W.; Palme, R.; Sachser, N. Activity changes and marked stereotypic behavior precede Abeta pathology in TgCRND8 Alzheimer mice. Neurobiol. Aging 2006, 27, 955–964. [Google Scholar] [CrossRef]
- Kang, J.E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef] [Green Version]
- Rothman, S.M.; Herdener, N.; Frankola, K.A.; Mughal, M.R.; Mattson, M.P. Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Abeta and pTau in a mouse model of Alzheimer’s disease. Brain Res. 2013, 1529, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Koh, K.; Zheng, X.; Sehgal, A. JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science 2006, 312, 1809–1812. [Google Scholar] [CrossRef] [Green Version]
- Hardin, P.E.; Panda, S. Circadian timekeeping and output mechanisms in animals. Curr. Opin. Neurobiol. 2013, 23, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Kress, G.J.; Liao, F.; Dimitry, J.; Cedeno, M.R.; FitzGerald, G.A.; Holtzman, D.M.; Musiek, E.S. Regulation of amyloid-beta dynamics and pathology by the circadian clock. J. Exp. Med. 2018, 215, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musiek, E.S.; Lim, M.M.; Yang, G.; Bauer, A.Q.; Qi, L.; Lee, Y.; Roh, J.H.; Ortiz-Gonzalez, X.; Dearborn, J.T.; Culver, J.P.; et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Investig. 2013, 123, 5389–5400. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Shi, Z.; Qin, C.; Tao, L.; Liu, X.; Xu, F.; Zhang, L.; Song, Y.; Liu, X.; Zhang, J. Therapeutic target database update 2012: A resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012, 40, D1128–D1136. [Google Scholar] [CrossRef]
- Hernandez-Boussard, T.; Whirl-Carrillo, M.; Hebert, J.M.; Gong, L.; Owen, R.; Gong, M.; Gor, W.; Liu, F.; Truong, C.; Whaley, R. The pharmacogenetics and pharmacogenomics knowledge base: Accentuating the knowledge. Nucleic Acids Res. 2007, 36, D913–D918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [Green Version]
- Pawson, A.J.; Sharman, J.L.; Benson, H.E.; Faccenda, E.; Alexander, S.P.; Buneman, O.P.; Davenport, A.P.; McGrath, J.C.; Peters, J.A.; Southan, C. The IUPHAR/BPS Guide to PHARMACOLOGY: An expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014, 42, D1098–D1106. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.D.; Armstrong, J.F.; Faccenda, E.; Southan, C.; Alexander, S.P.; Davenport, A.P.; Pawson, A.J.; Spedding, M.; Davies, J.A. The IUPHAR/BPS guide to PHARMACOLOGY in 2022: Curating pharmacology for COVID-19, malaria and antibacterials. Nucleic Acids Res. 2021, 50, D1282–D1294. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Wu, Z.; Wang, X.; Zhang, C.; Li, J.; Liu, G.; Tang, Y. Prediction of polypharmacological profiles of drugs by the integration of chemical, side effect, and therapeutic space. J. Chem. Inf. Modeling 2013, 53, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative toxicogenomics database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef] [PubMed]
- Tatonetti, N.P.; Patrick, P.Y.; Daneshjou, R.; Altman, R.B. Data-driven prediction of drug effects and interactions. Sci. Transl. Med. 2012, 4, 125-ra31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M.; Campillos, M.; Letunic, I.; Jensen, L.J.; Bork, P. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol. 2010, 6, 343. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Patel, C.J. A standard database for drug repositioning. Sci. Data 2017, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipscomb, C.E. Medical subject headings (MeSH). Bull. Med Libr. Assoc. 2000, 88, 265. [Google Scholar]
- Bodenreider, O. The unified medical language system (UMLS): Integrating biomedical terminology. Nucleic Acids Res. 2004, 32, D267–D270. [Google Scholar] [CrossRef] [Green Version]
- Chung, N.C.; Miasojedow, B.; Startek, M.; Gambin, A. Jaccard/Tanimoto similarity test and estimation methods for biological presence-absence data. BMC Bioinform. 2019, 20, 1–11. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Wang, J.Z.; Du, Z.; Payattakool, R.; Yu, P.S.; Chen, C.-F. A new method to measure the semantic similarity of GO terms. Bioinformatics 2007, 23, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Li, F.; Qin, Y.; Bo, X.; Wu, Y.; Wang, S. GOSemSim: An R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 2010, 26, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zeng, W.-M.; Cai, Y.-D.; Feng, K.-Y.; Chou, K.-C. Predicting anatomical therapeutic chemical (ATC) classification of drugs by integrating chemical-chemical interactions and similarities. PLoS ONE 2012, 7, e35254. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.; Britt, H. A new drug classification for computer systems: The ATC extension code. Int. J. Bio-Med. Comput. 1995, 40, 121–124. [Google Scholar] [CrossRef]
- Cao, S.; Lu, W.; Xu, Q. Deep neural networks for learning graph representations. In Proceedings of the AAAI Conference on Artificial Intelligence, Phoenix, AZ, USA, 12–17 February 2016. [Google Scholar]
- Gligorijević, V.; Barot, M.; Bonneau, R. deepNF: Deep network fusion for protein function prediction. Bioinformatics 2018, 34, 3873–3881. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Marin-Garcia, P.; Ping, P.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome diagram viewer: Data structures and strategies to boost performance. Bioinformatics 2018, 34, 1208–1214. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [Green Version]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef] [PubMed]
- Al Rihani, S.B.; Deodhar, M.; Darakjian, L.I.; Dow, P.; Smith, M.K.; Bikmetov, R.; Turgeon, J.; Michaud, V. Quantifying Anticholinergic Burden and Sedative Load in Older Adults with Polypharmacy: A Systematic Review of Risk Scales and Models. Drugs Aging 2021, 38, 977–994. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Agostini, J.V.; Allore, H.G. Cumulative anticholinergic exposure is associated with poor memory and executive function in older men. J. Am. Geriatr. Soc. 2008, 56, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Linjakumpu, T.; Hartikainen, S.; Klaukka, T.; Koponen, H.; Kivelä, S.-L. A model to classify the sedative load of drugs. Int. J. Geriatr. Psychiatry 2003, 18, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Boustani, M.; Campbell, N.; Munger, S.; Maidment, I.; Fox, C. Impact of anticholinergics on the aging brain: A review and practical application. Aging Health 2008, 4, 311–320. [Google Scholar] [CrossRef]
- Kiesel, E.K.; Hopf, Y.M.; Drey, M. An anticholinergic burden score for German prescribers: Score development. BMC Geriatr. 2018, 18, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jun, K.; Hwang, S.; Ah, Y.M.; Suh, Y.; Lee, J.Y. Development of an anticholinergic burden scale specific for Korean older adults. Geriatr. Gerontol. Int. 2019, 19, 628–634. [Google Scholar] [CrossRef]
- Sloane, P.; Ivey, J.; Roth, M.; Roederer, M.; Williams, C.S. Accounting for the sedative and analgesic effects of medication changes during patient participation in clinical research studies: Measurement development and application to a sample of institutionalized geriatric patients. Contemp. Clin. Trials 2008, 29, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, J.L.; Salow, M.J.; Angelini, M.C.; McGlinchey, R.E. The anticholinergic risk scale and anticholinergic adverse effects in older persons. Arch. Intern. Med. 2008, 168, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Sittironnarit, G.; Ames, D.; Bush, A.I.; Faux, N.; Flicker, L.; Foster, J.; Hilmer, S.; Lautenschlager, N.T.; Maruff, P.; Masters, C.L. Effects of anticholinergic drugs on cognitive function in older Australians: Results from the AIBL study. Dement. Geriatr. Cogn. Disord. 2011, 31, 173–178. [Google Scholar] [CrossRef]
- Nery, R.T.; Reis, A.M.M. Development of a Brazilian anticholinergic activity drug scale. Einstein 2019, 17, eAO4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kable, A.; Fullerton, A.; Fraser, S.; Palazzi, K.; Hullick, C.; Oldmeadow, C.; Pond, D.; Searles, A.; Edmunds, K.; Attia, J. Comparison of potentially inappropriate medications for people with dementia at admission and discharge during an unplanned admission to hospital: Results from the SMS dementia study. Healthcare 2019, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briet, J.; Javelot, H.; Heitzmann, E.; Weiner, L.; Lameira, C.; D’Athis, P.; Corneloup, M.; Vailleau, J.-L. The anticholinergic impregnation scale: Towards the elaboration of a scale adapted to prescriptions in French psychiatric settings. Therapies 2017, 72, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Klamer, T.T.; Wauters, M.; Azermai, M.; Durán, C.; Christiaens, T.; Elseviers, M.; Vander Stichele, R. A novel scale linking potency and dosage to estimate anticholinergic exposure in older adults: The muscarinic acetylcholinergic receptor ANTagonist exposure scale. Basic Clin. Pharmacol. Toxicol. 2017, 120, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Durán, C.E.; Azermai, M.; Vander Stichele, R.H. Systematic review of anticholinergic risk scales in older adults. Eur. J. Clin. Pharmacol. 2013, 69, 1485–1496. [Google Scholar] [CrossRef] [PubMed]
- Carnahan, R.M.; Lund, B.C.; Perry, P.J.; Pollock, B.G.; Culp, K.R. The anticholinergic drug scale as a measure of drug-related anticholinergic burden: Associations with serum anticholinergic activity. J. Clin. Pharmacol. 2006, 46, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Ehrt, U.; Broich, K.; Larsen, J.P.; Ballard, C.; Aarsland, D. Use of drugs with anticholinergic effect and impact on cognition in Parkinson’s disease: A cohort study. J. Neurol. Neurosurg. Psychiatry 2010, 81, 160–165. [Google Scholar] [CrossRef]
- Meguro, K.; Meguro, M.; Tanaka, Y.; Akanuma, K.; Yamaguchi, K.; Itoh, M. Risperidone is effective for wandering and disturbed sleep/wake patterns in Alzheimer’s disease. J. Geriatr. Psychiatry Neurol. 2004, 17, 61–67. [Google Scholar] [CrossRef]
- Cherukalady, R.; Kumar, D.; Basu, P.; Singaravel, M. Risperidone resets the circadian clock in mice. Biol. Rhythm. Res. 2017, 48, 583–591. [Google Scholar] [CrossRef]
- Omori, Y.; Kanbayashi, T.; Sagawa, Y.; Imanishi, A.; Tsutsui, K.; Takahashi, Y.; Takeshima, M.; Takaki, M.; Nishino, S.; Shimizu, T. Low dose of aripiprazole advanced sleep rhythm and reduced nocturnal sleep time in the patients with delayed sleep phase syndrome: An open-labeled clinical observation. Neuropsychiatr. Dis. Treat. 2018, 14, 1281. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, T. Improvement of a patient’s circadian rhythm sleep disorders by aripiprazole was associated with stabilization of his bipolar illness. J. Sleep Res. 2017, 26, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipchura, D.A.; Freyberg, Z.; Edwards, C.; Leckband, S.G.; McCarthy, M.J. Does the time of drug administration alter the metabolic risk of aripiprazole? Front. Psychiatry 2018, 9, 494. [Google Scholar] [CrossRef]
- Hirota, T.; Lewis, W.G.; Liu, A.C.; Lee, J.W.; Schultz, P.G.; Kay, S.A. A chemical biology approach reveals period shortening of the mammalian circadian clock by specific inhibition of GSK-3β. Proc. Natl. Acad. Sci. USA 2008, 105, 20746–20751. [Google Scholar] [CrossRef] [Green Version]
- Rock, P.L.; Goodwin, G.M.; Wulff, K.; McTavish, S.F.; Harmer, C.J. Effects of short-term quetiapine treatment on emotional processing, sleep and circadian rhythms. J. Psychopharmacol. 2016, 30, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, 363, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jiang, W.; Zhang, E.E. Orexin signaling regulates both the hippocampal clock and the circadian oscillation of Alzheimer’s disease-risk genes. Sci. Rep. 2016, 6, 36035. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.H.; Jiang, H.; Finn, M.B.; Stewart, F.R.; Mahan, T.E.; Cirrito, J.R.; Heda, A.; Snider, B.J.; Li, M.; Yanagisawa, M.; et al. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J. Exp. Med. 2014, 211, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Grippe, T.C.; Gonçalves, B.S.; Louzada, L.L.; Quintas, J.L.; Naves, J.O.; Camargos, E.F.; Nóbrega, O.T. Circadian rhythm in Alzheimer disease after trazodone use. Chronobiol. Int. 2015, 32, 1311–1314. [Google Scholar] [CrossRef]
- Ashford, J.W. Treatment of Alzheimer’s Disease: Trazodone, Sleep, Serotonin, Norepinephrine, and Future Directions. J. Alzheimer’s Dis. 2019, 67, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T. Distribution of Alzheimer’s neurofibrillary changes in the brain stem and hypothalamus of senile dementia. Acta Neuropathol. 1966, 6, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. Neurochemical studies of Alzheimer’s disease. Neurodegeneration 1996, 5, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Gonçalo, A.M.G.; Vieira-Coelho, M.A. The effects of trazodone on human cognition: A systematic review. Eur. J. Clin. Pharmacol. 2021, 77, 1623–1637. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Ellett, L.M.K.; Widagdo, I.S.; Pratt, N.L.; Roughead, E.E. Analysis of anticholinergic and sedative medicine effects on physical function, cognitive function, appetite and frailty: A cross-sectional study in Australia. BMJ Open 2019, 9, e029221. [Google Scholar] [CrossRef] [Green Version]
- van der Meer, H.G.; Wouters, H.; Pont, L.G.; Taxis, K. Reducing the anticholinergic and sedative load in older patients on polypharmacy by pharmacist-led medication review: A randomised controlled trial. BMJ Open 2018, 8, e019042. [Google Scholar] [CrossRef] [Green Version]
- Wouters, H.; van der Meer, H.; Taxis, K. Quantification of anticholinergic and sedative drug load with the Drug Burden Index: A review of outcomes and methodological quality of studies. Eur. J. Clin. Pharmacol. 2017, 73, 257–266. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Anticholinergic Burden | Sedative Load |
|---|---|---|
| Quetiapine | Moderate [76] | Moderate [77] |
| Aripiprazole | Low [78,79,80] | Moderate [81] |
| Risperidone | Low [76,78,79,80,82,83,84,85,86,87] | Moderate [77] |
| Suvorexant | No | High [77] |
| Olanzapine | Moderate [76,82] | Moderate [77] |
| Travoprost | No [79] | No |
| Betaxolol | Low [76,79,88] | Low [77] |
| Ibuprofen | No [79,80,89] | Low [77] |
| Trifluoperazine | High [78,82] | High [77] |
| Trazodone | Low [76,78,79,80,82,84,86,87] | Moderate [77] |
| Doxepin | High [78,79,83,89,90] | Moderate [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chyr, J.; Gong, H.; Zhou, X. DOTA: Deep Learning Optimal Transport Approach to Advance Drug Repositioning for Alzheimer’s Disease. Biomolecules 2022, 12, 196. https://doi.org/10.3390/biom12020196
Chyr J, Gong H, Zhou X. DOTA: Deep Learning Optimal Transport Approach to Advance Drug Repositioning for Alzheimer’s Disease. Biomolecules. 2022; 12(2):196. https://doi.org/10.3390/biom12020196
Chicago/Turabian StyleChyr, Jacqueline, Haoran Gong, and Xiaobo Zhou. 2022. "DOTA: Deep Learning Optimal Transport Approach to Advance Drug Repositioning for Alzheimer’s Disease" Biomolecules 12, no. 2: 196. https://doi.org/10.3390/biom12020196
APA StyleChyr, J., Gong, H., & Zhou, X. (2022). DOTA: Deep Learning Optimal Transport Approach to Advance Drug Repositioning for Alzheimer’s Disease. Biomolecules, 12(2), 196. https://doi.org/10.3390/biom12020196