
Morusflavone, a New Therapeutic Candidate for Prostate Cancer by CYP17A1 Inhibition: Exhibited by Molecular Docking and Dynamics Simulation

 ,
,  , ,
, ,
Abstract
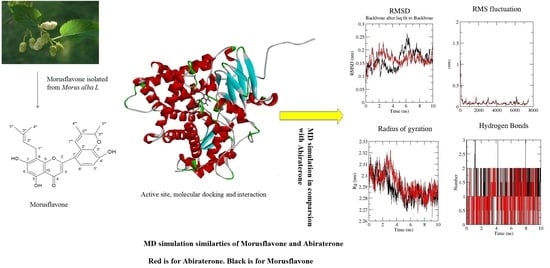
:
1. Introduction
2. Results
2.1. Molecular Docking
2.2. Trajectory Analysis of the MD Simulation
2.3. ADME and Toxicity Analysis
3. Discussion
4. Materials and Methods
4.1. Extraction, Isolation and Structure Elucidation
4.2. Molecular Docking
4.3. Molecular Dynamics Simulation Analysis
4.4. ADME and Toxicity Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varier, A. Dictionary of Indian Raw Materials & Industrial Products; Publication and Information Directorate Council of Scientific & Industrial Research: New Delhi, India, 2002; p. 387. [Google Scholar]
- Weiguo, Z.; Zhihua, Z.; Xuexia, M.; Yong, Z.; Sibao, W.; Jianhua, X.; Hui, P.; Yile, H.; Yongping, A. Comparison of genetic variation among wild and cultivated Morus Species (Moraceae: Morus) as revealed by ISSR and SSR markers. Biodivers. Conserv. 2007, 16, 275–290. [Google Scholar] [CrossRef]
- Ali, A.; Ali, M. Isolation and structure elucidation of a new linoleiyl glycoside and flavones from the stem bark of Morus alba L. Future J. Pharm. Sci. 2016, 2, 82–86. [Google Scholar] [CrossRef]
- Ali, A.; Ali, M. New triterpenoids from Morus alba L. stem bark. Nat. Prod. Res. 2013, 27, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ali, M. Phenyl alcohol and phenolic glycosides from the stem bark of Morus alba L. Nat. Prod. J. 2014, 4, 259–262. [Google Scholar] [CrossRef]
- Boszormenyi, A.; Szarka, S.z.; Hethelyi, E.; Gyurjan, I.; Laszlo, M.; Simandi, B.; Szoke, E.; Lemberkovics, E. Triterpenes in traditional and supercritical-fluid extracts of Morus alba leaf and stem bark. Acta Chromatogr. 2009, 21, 659–669. [Google Scholar] [CrossRef]
- Chen, C.; Mohamad Razali, U.H.; Saikim, F.H.; Mahyudin, A.; Mohd Noor, N.Q.I. Morus alba L. Plant: Bioactive compounds and potential as a functional food ingredient. Foods 2021, 10, 689. [Google Scholar] [CrossRef]
- Butt, M.S.; Nazir, A.; Sultan, M.T.; Schroen, K. Morus alba L. nature’s functional tonic. Trends Food. Sci. Technol. 2008, 19, 505–512. [Google Scholar] [CrossRef]
- Chan, E.W.C.; Wong, S.K.; Tangah, J.; Inoue, T.; Chan, H.T. Phenolic constituents and anticancer properties of Morus alba (white mulberry) leaves. J. Integr. Med. 2020, 18, 189–195. [Google Scholar] [CrossRef]
- Ha, M.T.; Tran, M.H.; Ah, K.J.; Jo, K.J.; Kim, J.; Kim, W.D.; Min, B.S. Potential pancreatic lipase inhibitory activity of phenolic constituents from the root bark of Morus alba L. Bioorg. Med. Chem. Lett. 2016, 26, 2788–2794. [Google Scholar] [CrossRef]
- Dat, N.T.; Binh, P.T.X.; Quynh, L.T.P.; Minh, C.V.; Huong, H.T.; Lee, J.J. Cytotoxic prenylated flavonoids from Morus alba. Fitoterapia 2010, 81, 1224–1227. [Google Scholar] [CrossRef]
- Jacobs, D.I.; Snoeijer, W.; Hallard, D.; Verpoorte, R. The Catharanthus alkaloids: Pharmacognosy and biotechnology. Curr. Med. Chem. 2004, 11, 607–628. [Google Scholar] [CrossRef]
- Huggins, C.; Stevens, R.E.; Hodges, C.V. Studies on prostatic cancer: II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941, 43, 209–223. [Google Scholar] [CrossRef]
- Goldberg, T.; Berrios-Colon, E. Abiraterone (zytiga), a novel agent for the management of castration-resistant prostate cancer. Pharm. Ther. 2013, 38, 23–26. [Google Scholar]
- Gomez, L.; Kovac, J.R.; Lamb, D.J. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids 2015, 95, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.W.; Comber, H.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2018. National Cancer Institute: Bethesda, MD, USA. Available online: https://seer.cancer.gov/csr/1975_2018/ (accessed on 20 July 2021).
- Rau, K.M.; Kang, H.Y.; Cha, T.L.; Miller, S.A.; Hung, M.C. The mechanisms and managements of hormone-therapy resistance in breast and prostate cancers. Endocr.-Relat. Cancer 2005, 12, 511–532. [Google Scholar] [CrossRef] [Green Version]
- Attard, G.; Cooper, C.S.; de Bono, J.S. Steroid hormone receptors in prostate cancer: A hard habit to break? Cancer Cell 2009, 16, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Eo, H.J.; Park, J.H.; Park, G.H.; Lee, M.H.; Koo, J.R.; Jeong, J.S. Anti-inflammatory and anti-cancer activity of mulberry (Morus alba L.) root bark. BMC Complement. Altern. Med. 2014, 14, 200. [Google Scholar] [CrossRef] [Green Version]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Zhou, Y. Improving prediction of secondary structure, local backbone angles and solvent accessible surface area of proteins by iterative deep learning. Sci. Rep. 2015, 5, 11476. [Google Scholar] [CrossRef] [Green Version]
- Trisciuzzi, D.; Nicolotti, O.; Miteva, M.A.; Villoutreix, B.O. Analysis of solvent-exposed and buried co-crystallized ligands: A case study to support the design of novel protein–protein interaction inhibitors. Drug Dis. Today 2019, 24, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Balani, S.K.; Miwa, G.T.; Gan, L.S.; Wu, J.T.; Lee, F.W. Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection. Curr. Top. Med. Chem. 2005, 5, 1033–1038. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Auto-Dock4 and Auto-DockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Hallida, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Abdi, S.A.H.; Alzahrani, A.; Asad, M.; Alquraini, A.; Alghamdi, A.I.; Sayed, S.F. Molecular docking and dynamics simulation to screen interactive potency and stability of fungicide tebuconazole with thyroid and sex hormone-binding globulin: Implications of endocrine and reproductive interruptions. J. Appl. Toxicol. 2021. [Google Scholar] [CrossRef]
- Lee, H.C.; Hsu, W.C.; Liu, A.L.; Hsu, C.J.; Sun, Y.C. Using thermodynamic integration MD simulation to compute relative protein– ligand binding free energy of a GSK3β kinase inhibitor and its analogs. J. Mol. Graph. Model. 2014, 51, 37–49. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Tang, Y. AdmetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | H-Bonds | H Bond Distance (Å) | Amino Acid Residues Involved in Hydrophobic Interactions | Docking Final Intermolecular Energy (ΔG) = vdW + Hbond + Desolv Energy (Kcal/mol) | Inhibition Constant (Ki) | |
|---|---|---|---|---|---|---|
| Between Hydrogen and Acceptor Atom | Between Donor and Acceptor Atom | |||||
| Morusflavone | ARG 125 | 3.01 | 3.53 | PHE 114 | −10.3 | 103.87 nM |
| ILE 443 | 1.76 | 2.60 | ALA 302 | |||
| VAL 482 | 2.03 | 2.75 | ILE 443 | |||
| VAL 482 | ||||||
| Abiraterone | ILE 443 | 3.01 | 3.71 | ILE 443 | −10.07 | 174.43 nM |
| GLY 444 | 2.31 | 2.85 | ALA 113 302, 367 | |||
| CYS 442 | 2.18 | 2.86 | PHE 114 | |||
| VAL 482 | ||||||
| Enzyme Substrate Complex | RMSD (nm) | RMSF (nm) | RG (nm) | SASA (nm2) | Interaction Energy (KJ/mol) |
|---|---|---|---|---|---|
| Morusflavone–CYP17A1 | 0.05 ± 0.02 | 0.25 ± 0.01 | 2.29 ± 0.02 | 7.25 ± 0.01 | –246.252 |
| Abiraterone–CYP17A1 | 0.05 ± 0.02 | 0.25 ± 0.01 | 2.31 ± 0.02 | 6.6 ± 0.01 | –207.86 |
| S. No. | Parameters | Results |
|---|---|---|
| 1 | Lipinski violation | No |
| 2 | No. of H-bond acceptors | 6 |
| 3 | No. of H-bond donors | 3 |
| 4 | Molecular weight | 420.45 g/mol |
| 5 | GI absorption | High |
| 6 | BBB permeation | No |
| 7 | CYP1A2 inhibitor | No |
| 8 | CYP2C19 inhibitor | No |
| 9 | CYP2C9 inhibitor | Yes |
| 10 | Log Kp (skin permeation) | −5.14 cm/s |
| 11 | Acute Oral Toxicity (c) | Category III |
| 12 | Eye irritation | No |
| 13 | Ames mutagenesis | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdi, S.A.H.; Ali, A.; Sayed, S.F.; Ahsan, M.J.; Tahir, A.; Ahmad, W.; Shukla, S.; Ali, A. Morusflavone, a New Therapeutic Candidate for Prostate Cancer by CYP17A1 Inhibition: Exhibited by Molecular Docking and Dynamics Simulation. Plants 2021, 10, 1912. https://doi.org/10.3390/plants10091912
Abdi SAH, Ali A, Sayed SF, Ahsan MJ, Tahir A, Ahmad W, Shukla S, Ali A. Morusflavone, a New Therapeutic Candidate for Prostate Cancer by CYP17A1 Inhibition: Exhibited by Molecular Docking and Dynamics Simulation. Plants. 2021; 10(9):1912. https://doi.org/10.3390/plants10091912
Chicago/Turabian StyleAbdi, Sayed Aliul Hasan, Amena Ali, Shabihul Fatma Sayed, Mohamed Jawed Ahsan, Abu Tahir, Wasim Ahmad, Shatrunajay Shukla, and Abuzer Ali. 2021. "Morusflavone, a New Therapeutic Candidate for Prostate Cancer by CYP17A1 Inhibition: Exhibited by Molecular Docking and Dynamics Simulation" Plants 10, no. 9: 1912. https://doi.org/10.3390/plants10091912
APA StyleAbdi, S. A. H., Ali, A., Sayed, S. F., Ahsan, M. J., Tahir, A., Ahmad, W., Shukla, S., & Ali, A. (2021). Morusflavone, a New Therapeutic Candidate for Prostate Cancer by CYP17A1 Inhibition: Exhibited by Molecular Docking and Dynamics Simulation. Plants, 10(9), 1912. https://doi.org/10.3390/plants10091912