
Targeted Whole Genome Sequencing (TWG-Seq) of Cucumber Green Mottle Mosaic Virus Using Tiled Amplicon Multiplex PCR and Nanopore Sequencing
 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Relative Quantification of Viral Load
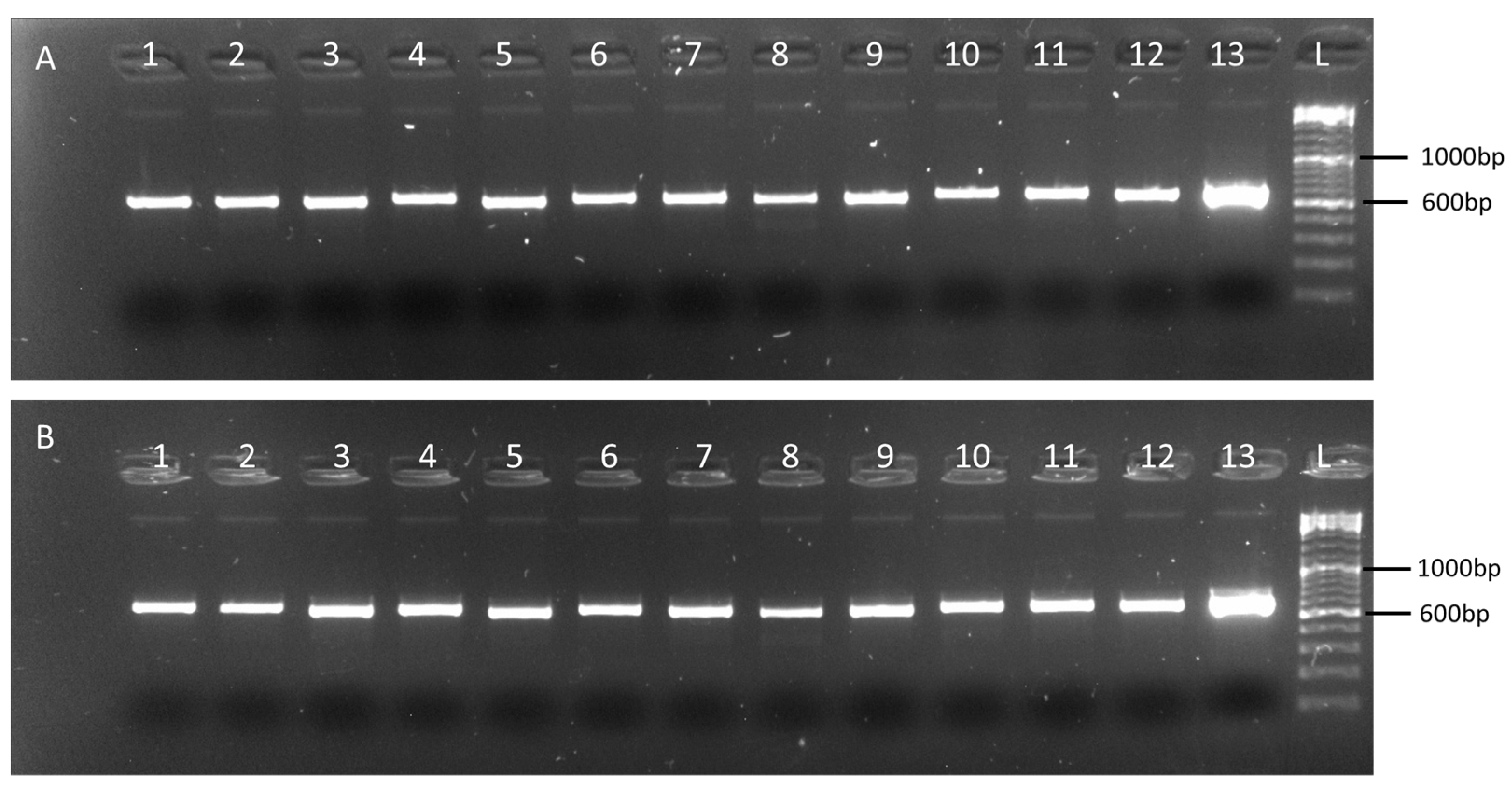
2.2. Validation of the Multiplex Tiling PCR Assays
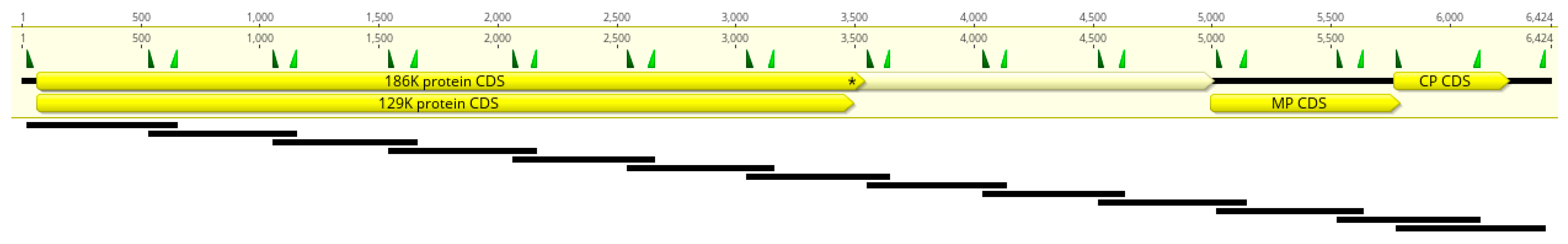
2.3. Primer Scheme Analysis
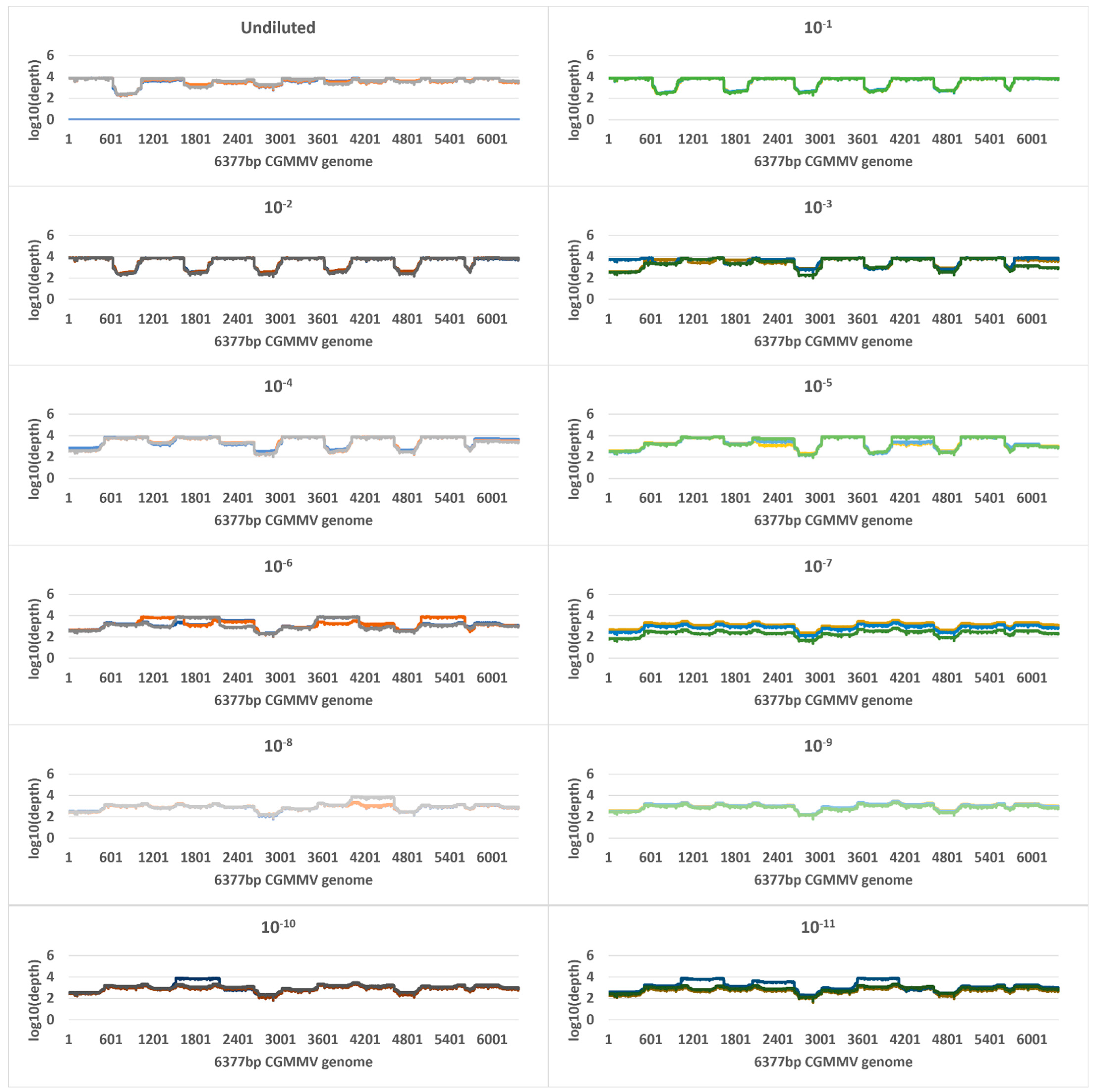
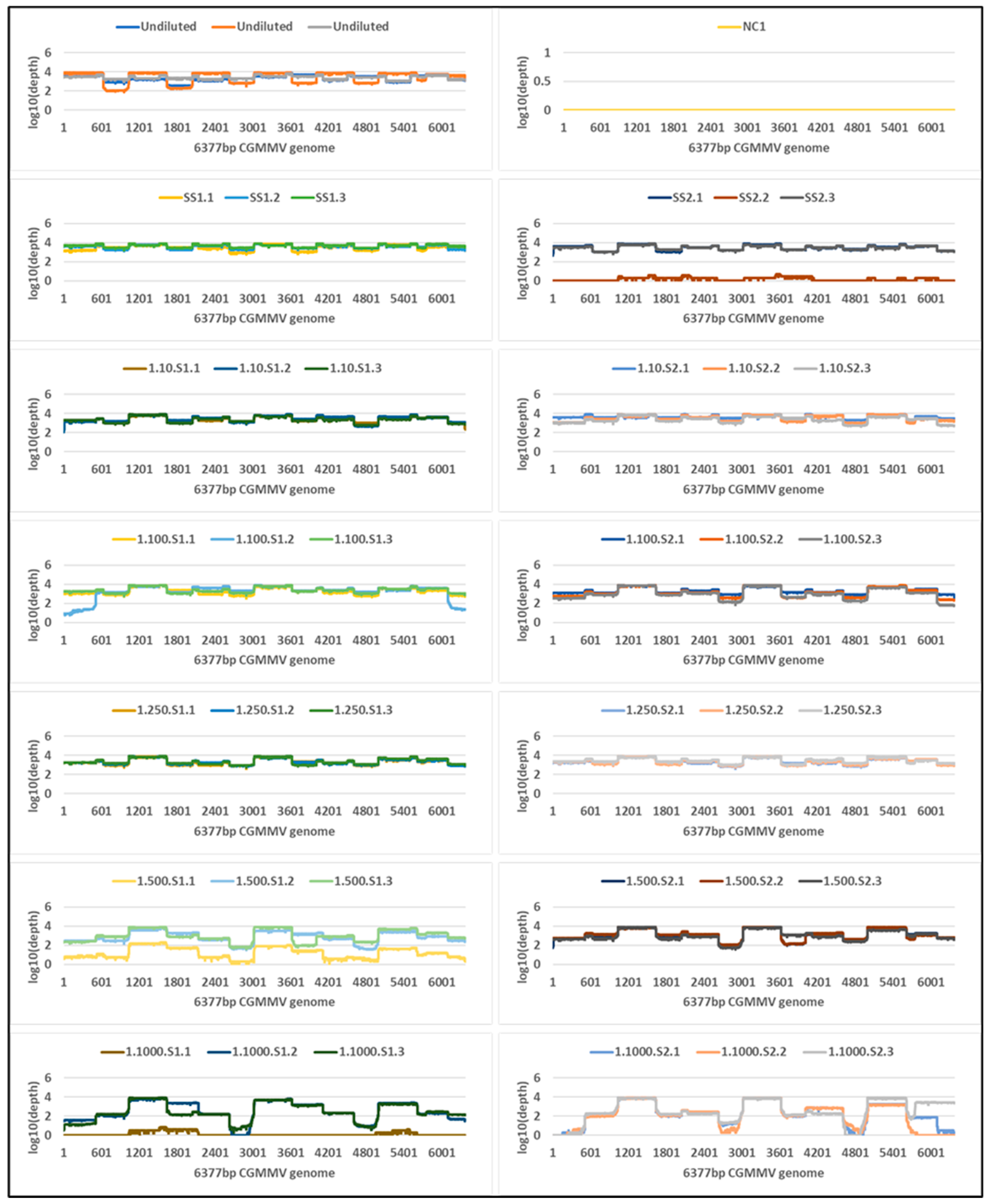
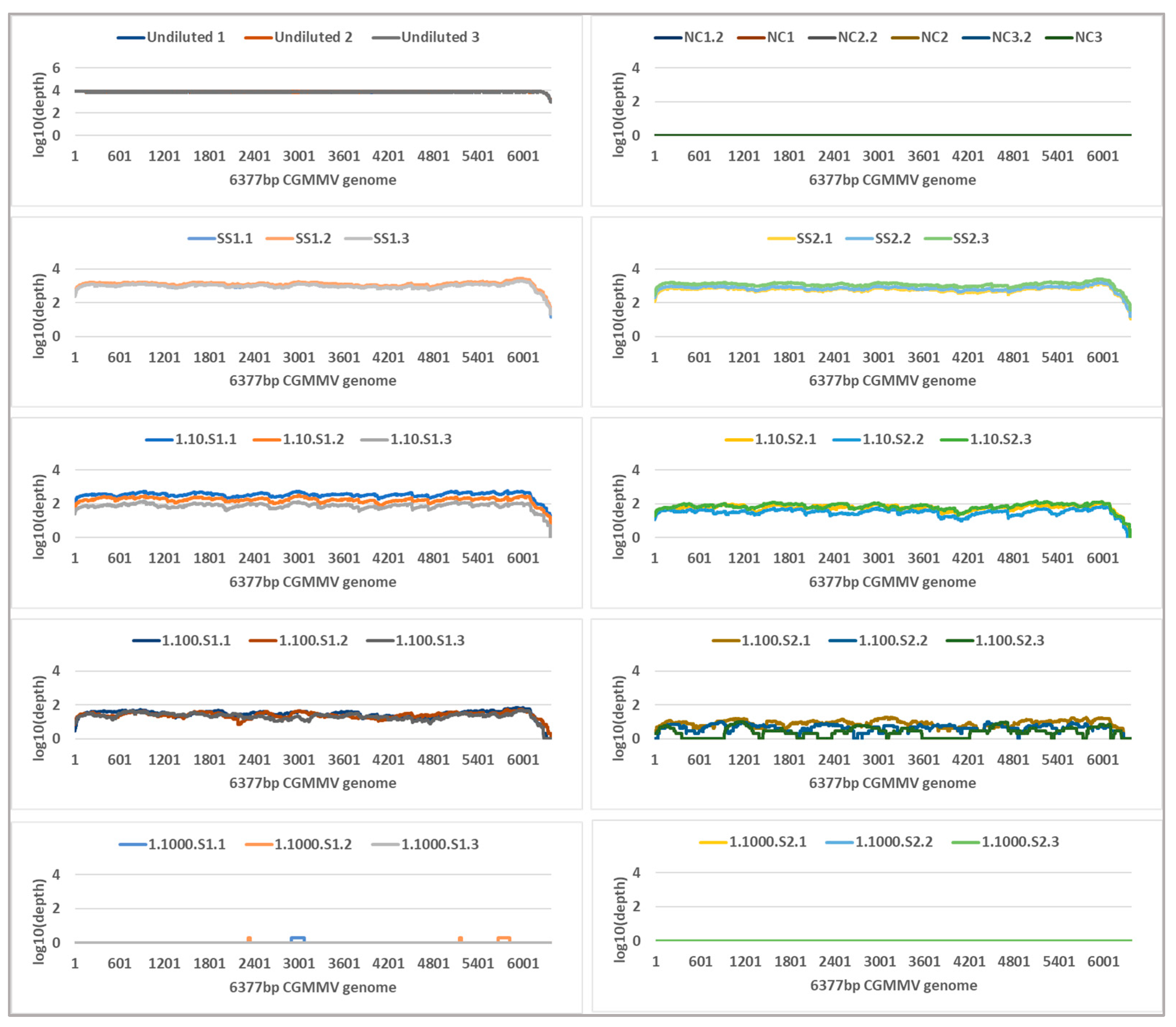
2.4. Tiled Amplicon Sequencing
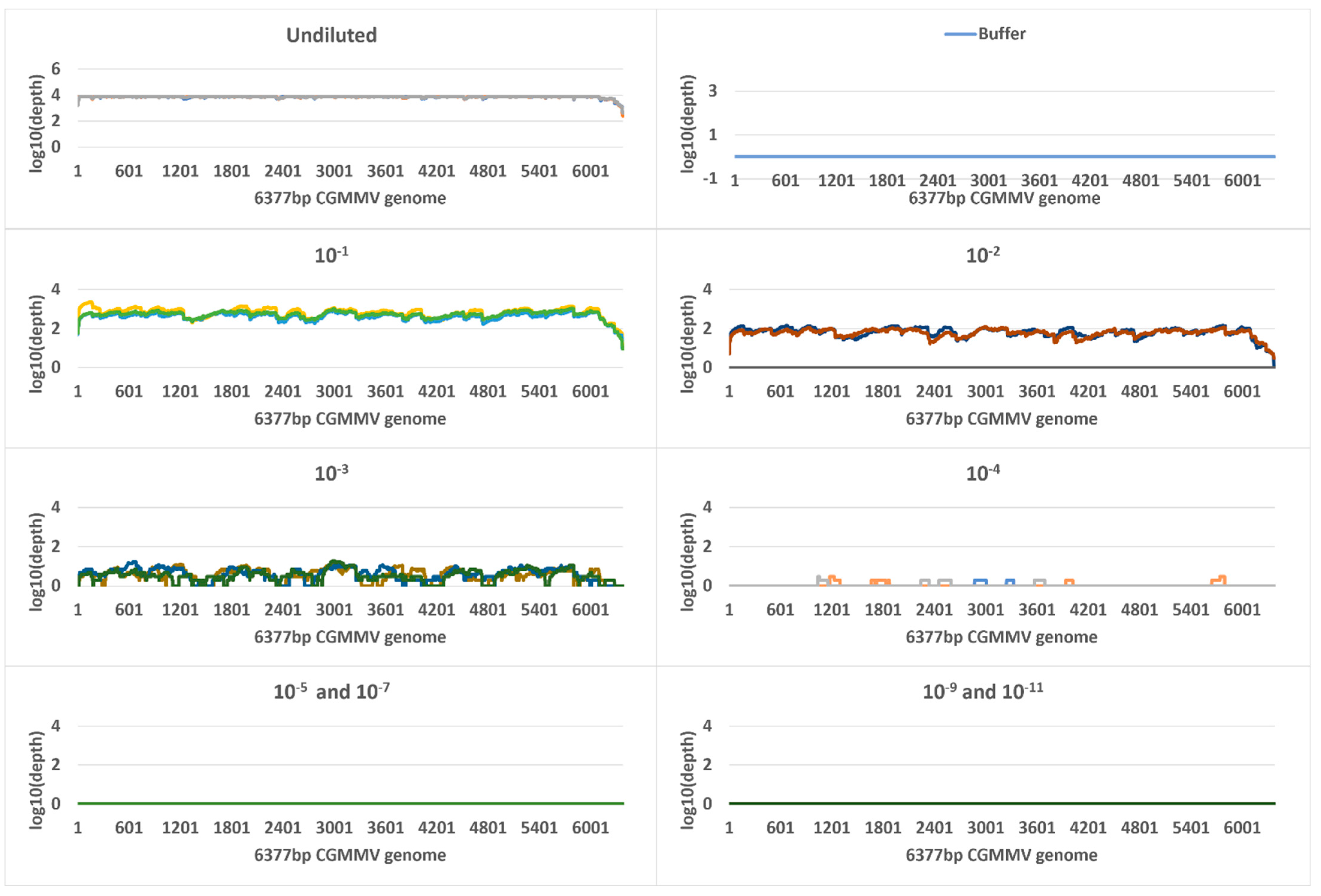
2.5. Metagenomic Sequencing
2.6. Sequence Identity Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Preparation of Virus Panels
4.3. RT-qPCR Conditions
4.4. Transcribed RNA Standards for Absolute Quantification of CGMMV by RT-qPCR
4.5. Multiplex PCR Primer Design
4.6. Multiplex Tiling PCR
4.7. Tiled Amplicon Sequencing and Bioinformatics
4.8. Metagenomic Sequencing and Bioinformatics
4.9. Sequence Accuracy Assessment
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tesoriero, L.A.; Chambers, G.; Srivastava, M.; Smith, S.; Conde, B.; Tran-Nguyen, L.T.T. First Report of Cucumber Green Mottle Mosaic Virus in Australia. Australas. Plant Dis. Notes 2015, 11, 1. [Google Scholar] [CrossRef] [Green Version]
- Lovelock, D.; Mintoff, S.; Kurz, N.; Neilsen, M.; Patel, S.; Constable, F.; Tran-Nguyen, L. Investigating the Longevity and Infectivity of Cucumber Green Mottle Mosaic Virus in Soils of the Northern Territory, Australia. Plants 2022, 11, 883. [Google Scholar] [CrossRef] [PubMed]
- Dombrovsky, A.; Tran-Nguyen, L.T.T.; Jones, R.A.C. Cucumber Green Mottle Mosaic Virus: Rapidly Increasing Global Distribution, Etiology, Epidemiology, and Management. Annu. Rev. Phytopathol. 2017, 55, 231–256. [Google Scholar] [CrossRef] [PubMed]
- Tran-Nguyen, L. Cucumber Green Mottle Mosaic Virus in Australia: The Story so Far. In Proceedings of the 13th International Plant Virus Epidemiology Symposium, Avignon, France, 6–10 June 2016. [Google Scholar]
- Tran-Nguyen, L.T.T. Final Report VG15013 Improved Management Options for Cucumber Green Mottle Mosaic Virus; Horticulture Innovation Australia: Sydney, Australia, 2019; p. 106. ISBN 978-0-7341-4506-2. [Google Scholar]
- Kehoe, M.A.; Webster, C.; Wang, C.; Jones, R.A.C.; Coutts, B.A. Occurrence of Cucumber Green Mottle Mosaic Virus in Western Australia. Australas. Plant Pathol. 2022, 51, 1–8. [Google Scholar] [CrossRef]
- Constable, F.; Daly, A.; Terras, M.A.; Penrose, L.; Dall, D. Detection in Australia of Cucumber Green Mottle Mosaic Virus in Seed Lots of Cucurbit Crops. Australas. Plant Dis. Notes 2018, 13, 18. [Google Scholar] [CrossRef] [Green Version]
- Robinson, E.R.; Walker, T.M.; Pallen, M.J. Genomics and Outbreak Investigation: From Sequence to Consequence. Genome Med. 2013, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A.; Hewlett, E.L. Whole-Genome Sequencing in Outbreak Analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef] [Green Version]
- Fox, A.; Fowkes, A.R.; Skelton, A.; Harju, V.; Buxton-Kirk, A.; Kelly, M.; Forde, S.M.D.; Pufal, H.; Conyers, C.; Ward, R.; et al. Using High-Throughput Sequencing in Support of a Plant Health Outbreak Reveals Novel Viruses in Ullucus Tuberosus (Basellaceae). Plant Pathol. 2019, 68, 576–587. [Google Scholar] [CrossRef]
- Deng, X.; Achari, A.; Federman, S.; Yu, G.; Somasekar, S.; Bártolo, I.; Yagi, S.; Mbala-Kingebeni, P.; Kapetshi, J.; Ahuka-Mundeke, S.; et al. Metagenomic Sequencing with Spiked Primer Enrichment for Viral Diagnostics and Genomic Surveillance. Nat. Microbiol. 2020, 5, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Piombo, E.; Abdelfattah, A.; Droby, S.; Wisniewski, M.; Spadaro, D.; Schena, L. Metagenomics Approaches for the Detection and Surveillance of Emerging and Recurrent Plant Pathogens. Microorganisms 2021, 9, 188. [Google Scholar] [CrossRef]
- Hasiów-Jaroszewska, B.; Boezen, D.; Zwart, M.P. Metagenomic Studies of Viruses in Weeds and Wild Plants: A Powerful Approach to Characterise Variable Virus Communities. Viruses 2021, 13, 1939. [Google Scholar] [CrossRef]
- Tedersoo, L.; Drenkhan, R.; Anslan, S.; Morales-Rodriguez, C.; Cleary, M. High-Throughput Identification and Diagnostics of Pathogens and Pests: Overview and Practical Recommendations. Mol. Ecol. Resour. 2019, 19, 47–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardy, J.L.; Loman, N.J. Towards a Genomics-Informed, Real-Time, Global Pathogen Surveillance System. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Liefting, L.W.; Waite, D.W.; Thompson, J.R. Application of Oxford Nanopore Technology to Plant Virus Detection. Viruses 2021, 13, 1424. [Google Scholar] [CrossRef] [PubMed]
- Stobbe, A.H.; Roossinck, M.J. Plant Virus Metagenomics: What We Know and Why We Need to Know More. Front. Plant Sci. 2014, 5, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaafar, Y.Z.A.; Ziebell, H. Comparative Study on Three Viral Enrichment Approaches Based on RNA Extraction for Plant Virus/Viroid Detection Using High-Throughput Sequencing. PLoS ONE 2020, 15, e0237951. [Google Scholar] [CrossRef]
- Gaafar, Y.Z.A.; Westenberg, M.; Botermans, M.; László, K.; De Jonghe, K.; Foucart, Y.; Ferretti, L.; Kutnjak, D.; Pecman, A.; Mehle, N.; et al. Interlaboratory Comparison Study on Ribodepleted Total RNA High-Throughput Sequencing for Plant Virus Diagnostics and Bioinformatic Competence. Pathogens 2021, 10, 1174. [Google Scholar] [CrossRef]
- Espindola, A.S.; Sempertegui-Bayas, D.; Bravo-Padilla, D.F.; Freire-Zapata, V.; Ochoa-Corona, F.; Cardwell, K.F. TASPERT: Target-Specific Reverse Transcript Pools to Improve HTS Plant Virus Diagnostics. Viruses 2021, 13, 1223. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and Biological Insights from Viral Genome Sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Oliveira, B.B.; Veigas, B.; Baptista, P.V. Isothermal Amplification of Nucleic Acids: The Race for the Next “Gold Standard. ” Front. Sens. 2021, 2, 752600. [Google Scholar] [CrossRef]
- Worobey, M.; Watts, T.D.; McKay, R.A.; Suchard, M.A.; Granade, T.; Teuwen, D.E.; Koblin, B.A.; Heneine, W.; Lemey, P.; Jaffe, H.W. 1970s and ‘Patient 0’ HIV-1 Genomes Illuminate Early HIV/AIDS History in North America. Nature 2016, 539, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, A.; Ulm, S.-L.; Uddin, S.; Förster, S.; Seifert, D.; Oehme, R.; Corty, M.; Schaade, L.; Michel, J.; Nitsche, A. AmpliCoV: Rapid Whole-Genome Sequencing Using Multiplex PCR Amplification and Real-Time Oxford Nanopore MinION Sequencing Enables Rapid Variant Identification of SARS-CoV-2. Front. Microbiol. 2021, 12, 1703. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, S.C.B.; Blacklaws, B.A.; Yohan, B.; Yudhaputri, F.A.; Hayati, R.F.; Schwem, B.; Salvaña, E.M.; Destura, R.V.; Lester, J.S.; Myint, K.S.; et al. Assessment of a Multiplex PCR and Nanopore-Based Method for Dengue Virus Sequencing in Indonesia. Virol. J. 2020, 17, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Leung, A.W.-S.; Zheng, Z.; Zhang, D.; Xiao, C.; Luo, R.; Luo, M.; Zhang, S. Applications and Potentials of Nanopore Sequencing in the (Epi)Genome and (Epi)Transcriptome Era. Innov. 2021, 2, 100153. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, S.; Oosterhof, J. TaqMan Assays Designed on the Coding Sequence of the Movement Protein of Cucumber Green Mottle Mosaic Virus for Its Detection in Cucurbit Seeds. In Proceedings of the 2015 APS Annual Meeting, New York, NY, USA, 21–24 May 2015. [Google Scholar]
- Maina, S.; Zheng, L.; Rodoni, B.C. Targeted Genome Sequencing (TG-Seq) Approaches to Detect Plant Viruses. Viruses 2021, 13, 583. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC Multiplex PCR Method for SARS-CoV-2 Genome Sequencing Using Nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gardner, S.N.; Jaing, C.J.; Elsheikh, M.M.; Peña, J.; Hysom, D.A.; Borucki, M.K. Multiplex Degenerate Primer Design for Targeted Whole Genome Amplification of Many Viral Genomes. Adv. Bioinform. 2014, 2014, 101894. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Shrivastava, S.; Brownley, A.; Katzel, D.; Bera, J.; Nguyen, A.T.; Thovarai, V.; Halpin, R.; Stockwell, T.B. Automated Degenerate PCR Primer Design for High-Throughput Sequencing Improves Efficiency of Viral Sequencing. Virol. J. 2012, 9, 1–9. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Lin, Y.; Bo, X.; Song, H.; Li, K.; Li, P.; Ni, M. Assessment of Two-Pool Multiplex Long-Amplicon Nanopore Sequencing of SARS-CoV-2. J. Med. Virol. 2022, 94, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and Inexpensive Whole-Genome Sequencing of SARS-CoV-2 Using 1200 Bp Tiled Amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef] [PubMed]
- Kwok, S.; Kellogg, D.E.; McKinney, N.; Spasic, D.; Goda, L.; Levenson, C.; Sninsky, J.J. Effects of Primer-Template Mismatches on the Polymerase Chain Reaction: Human Immunodeficiency Virus Type 1 Model Studies. Nucleic Acids Res. 1990, 18, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, D.J.; McLean, M.A.; Mukerji, S.; Green, M. Improved RNA Extraction from Woody Plants for the Detection of Viral Pathogens by Reverse Transcription-Polymerase Chain Reaction. Plant Dis. 1997, 81, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Nancarrow, N.; Constable, F.E.; Finlay, K.J.; Freeman, A.J.; Rodoni, B.C.; Trebicki, P.; Vassiliadis, S.; Yen, A.L.; Luck, J.E. The Effect of Elevated Temperature on Barley Yellow Dwarf Virus-PAV in Wheat. Virus Res. 2014, 186, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega for Making Accurate Alignments of Many Protein Sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. In Proceedings of the 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, USA, 17–20 March 2014. [Google Scholar]
- Au, C.H.; Ho, D.N.; Kwong, A.; Chan, T.L.; Ma, E.S.K. BAMClipper: Removing Primers from Alignments to Minimize False-Negative Mutations in Amplicon Next-Generation Sequencing. Sci. Rep. 2017, 7, 1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F. Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 31 March 2021).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Cq | Virus Copies Per µL of RNA (SD) | Tiled Amplicon—MinION | Untargeted Metagenomic—NovaSeq | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reads Used | Reads Mapped to CGMMV (SD) | % Ref. Bases Covered | Average Read Depth | Reads Used | Reads Mapped to CGMMV (SD) | % Ref. Bases Covered | Average Read Depth | |||
| Undiluted a | 9.5 | 724,858,724 (505,575,547) | 119,982 | 119,737 (19,326) | 100 | 4633 | 7,948,784 | 705,257 (99,781) | 100 | 7535 |
| 10−1 | 11.8 | 148,495,975 (48,814,341) | 428,078 | 427,838 (39,232) | 100 | 5413 | 8,565,097 | 24,079 (5912) | 100 | 585 |
| 10−2 | 15 | 18,581,696 (4,560,628) | 285,974 | 285,786 (44,622) | 100 | 5331 | 10,941,741 | 2842 (1648) | 100 | 69 |
| 10−3 | 18 | 4,400,508 (5,288,129) | 177,081 | 176,916 (107,377) | 100 | 4432 | 10,444,239 | 185 (19) | 95 | 5 |
| 10−4 | 21.5 | 322,122 (174,102) | 220,367 | 220,147 (45,775) | 100 | 4301 | 10,212,900 | 15 (4) | 25 | 0 |
| 10−5 | 25.7 | 20,532 (3610) | 85,189 | 85,031 (29,087) | 100 | 3293 | 9,821,045 | 0 | 0 | 0 |
| 10−6 | 29 | 2316 (250) | 60,563 | 60,300 (14,479) | 100 | 2251 | 12,239,795 | 0 | 0 | 0 |
| 10−7 | 33.6 | 125 (26) | 9794 | 9433 (6603) | 100 | 893 | 15,300,104 | 0 | 0 | 0 |
| 10−8 | 37.1 | 15 (10) | 10,721 | 18,509 (15,356) | 100 | 1098 | 10,047,613 | 3 | 6.6 | 0.07 |
| 10−9 | 37.9 | 23 | 11,992 | 11,225 (1771) | 100 | 1060 | 7,751,478 | 0 | 0 | 0 |
| 10−10 | 37.3 | 17 (17) | 26,385 | 17,084 (10,639) | 100 | 1201 | 14,537,532 | 0 | 0 | 0 |
| 10−11 | 38 | 8 (4) | 8725 | 18,494 (18,593) | 100 | 1293 | 11,493,244 | 0 | 0 | 0 |
| Buffer b | 36.8 | 22 (21) | 1532 | 0 | 0 | 0 | 10,699,014 | 1 | 2.4 | 0.02 |
| PCR NTC c | NA | NA | 7 | 0 | 0 | 0 | NA | NA | NA | NA |
| Sample | Cq | Virus Copies Per µL of RNA (SD) | Tiled Amplicon—MinION | Untargeted Metagenomic—NovaSeq | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reads Used | Reads Mapped to CGMMV (SD) | % Ref Bases Covered | Average Read Depth | Reads Used | Reads Mapped to CGMMV (SD) | % Ref Bases Covered | Average Read Depth | |||
| Undiluted a | 8.4 | 1,526,567,296 (1,113,748,195) | 48,837 | 48,827 (34,308) | 100 | 3417 | 2,844,308 | 2,705,523 (246,410) | 100 | 7820 |
| Single seed 1 | 13.8 | 44,181,975 (3,663,819) | 61,585 | 61,543 (13,126) | 100 | 4284 | 6,586,247 | 28,232 (4263) | 100 | 1149 |
| Single seed 2 | 13.8 | 35,809,461 (3,057,510) | 26,005 | 25,996 (22,799) | 100 | 3445 | 4,967,016 | 21,700 (7948) | 100 | 873 |
| 1 seed in 10: Seed 1 | 18.1 | 3,016,299 (2,079,368) | 34,912 | 34,901 (4711) | 100 | 3029 | 5,231,106 | 4940 (3312) | 100 | 204 |
| 1 seed in 10: Seed 2 | 16.3 | 9,965,820 (7,389,633) | 67,942 | 67,919 (36,596) | 100 | 3837 | 8,859,498 | 1472 (423) | 100 | 57 |
| 1 seed in 100: Seed 1 | 20.9 | 429,383 (121,406) | 35,503 | 35,483 (9375) | 100 | 2847 | 9,101,950 | 684 (124) | 100 | 27 |
| 1 seed in 100: Seed 2 | 19.3 | 1,190,964 (216,733) | 42,308 | 42,295 (6359) | 100 | 2639 | 6,293,380 | 142 (79) | 94 | 6 |
| 1 seed in 250: Seed 1 | 21.4 | 331,757 (116,464) | 36,270 | 36,259 (4850) | 100 | 2871 | 5,577,062 | 298 (60) | 99 | 12 |
| 1 seed in 250: Seed 2 | 20.3 | 628,026 (195,273) | 50,390 | 50,377 (11,703) | 100 | 3224 | 4,846,547 | 209 (146) | 96 | 8 |
| 1 seed in 500: Seed 1 | 24.3 | 47,418 (6915) | 17,362 | 17,351 (17,552) | 100 | 1939 | 4,108,499 | 17 (9) | 45 | 1 |
| 1 seed in 500: Seed 2 | 24.4 | 46,624 (7676) | 50,839 | 50,823 (20,825) | 100 | 2565 | 4,298,383 | 28 (11) | 61 | 1 |
| 1 seed in 1000: Seed 1 | 30.4 | 947 (106) | 12,185 | 12,177 (10,585) | 98 | 1569 | 4,164,645 | 4 (2) | 17 | 0 |
| 1 seed in 1000: Seed 2 | 27.0 | 8894 (1410) | 39,969 | 39,958 (7833) | 95 | 2031 | 2,063,940 | 0 | 0 | 0 |
| NC1 b | UD | 0 | NA | NA | NA | NA | 5,672,805 | 0 | 0 | 0 |
| NC2 b | UD | 0 | NA | NA | NA | NA | 4,270,709 | 0 | 0 | 0 |
| PCR NTC c | NA | NA | 10,941 | 11 | 48 | 1 | NA | NA | NA | NA |
| Tiled Amplicon—MinION | Untargeted Metagenomic—NovaSeq | |||||||
|---|---|---|---|---|---|---|---|---|
| Seed Interception Isolate | Reads Used | Mapped Reads | Percentage of Reference Bases Covered | Average Read Depth | Reads Used | Mapped Reads | Percentage of Reference Bases Covered | Average Read Depth |
| Citrullus lanatus_2018-1 | 8701 | 8633 | 100 | 797.156 | 13,808,999 | 3603 | 99.16 | 85.3656 |
| Citrullus lanatus_2018-2 | 10,121 | 10,012 | 100 | 916.348 | 9,726,986 | 3858 | 99.21 | 90.6358 |
| Cucumis melo_2015-1 | 83,828 | 77,781 | 100 | 2646.63 | 5,335,679 | 291 | 80.49 | 5.86938 |
| Cucumis sativus_2014-1 | 1003 | 57 | 78.96 | 5.19711 | 1,652,058 | 14 | 11.38 | 0.265608 |
| Samples | Reads Used | Mapped Reads | Average Coverage with Deletion | Percentage of Reference Bases Covered |
|---|---|---|---|---|
| KGMMV-1 | 316 | 2 | 0.105 | 5.49 |
| KGMMV-2 | 213 | 1 | 0 | 8.04 |
| ZGMMV-1 | 1090 | 0 | 0 | 0 |
| ZGMMV-2 | 132 | 0 | 0 | 0 |
| NTC-1 | 106 | 0 | 0 | 0 |
| NTC-2 | 144 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mackie, J.; Kinoti, W.M.; Chahal, S.I.; Lovelock, D.A.; Campbell, P.R.; Tran-Nguyen, L.T.T.; Rodoni, B.C.; Constable, F.E. Targeted Whole Genome Sequencing (TWG-Seq) of Cucumber Green Mottle Mosaic Virus Using Tiled Amplicon Multiplex PCR and Nanopore Sequencing. Plants 2022, 11, 2716. https://doi.org/10.3390/plants11202716
Mackie J, Kinoti WM, Chahal SI, Lovelock DA, Campbell PR, Tran-Nguyen LTT, Rodoni BC, Constable FE. Targeted Whole Genome Sequencing (TWG-Seq) of Cucumber Green Mottle Mosaic Virus Using Tiled Amplicon Multiplex PCR and Nanopore Sequencing. Plants. 2022; 11(20):2716. https://doi.org/10.3390/plants11202716
Chicago/Turabian StyleMackie, Joanne, Wycliff M. Kinoti, Sumit I. Chahal, David A. Lovelock, Paul R. Campbell, Lucy T. T. Tran-Nguyen, Brendan C. Rodoni, and Fiona E. Constable. 2022. "Targeted Whole Genome Sequencing (TWG-Seq) of Cucumber Green Mottle Mosaic Virus Using Tiled Amplicon Multiplex PCR and Nanopore Sequencing" Plants 11, no. 20: 2716. https://doi.org/10.3390/plants11202716
APA StyleMackie, J., Kinoti, W. M., Chahal, S. I., Lovelock, D. A., Campbell, P. R., Tran-Nguyen, L. T. T., Rodoni, B. C., & Constable, F. E. (2022). Targeted Whole Genome Sequencing (TWG-Seq) of Cucumber Green Mottle Mosaic Virus Using Tiled Amplicon Multiplex PCR and Nanopore Sequencing. Plants, 11(20), 2716. https://doi.org/10.3390/plants11202716