
Genome-Wide Association Reveals Trait Loci for Seed Glucosinolate Accumulation in Indian Mustard (Brassica juncea L.)


Abstract
:1. Introduction
2. Results
2.1. Genotype Data
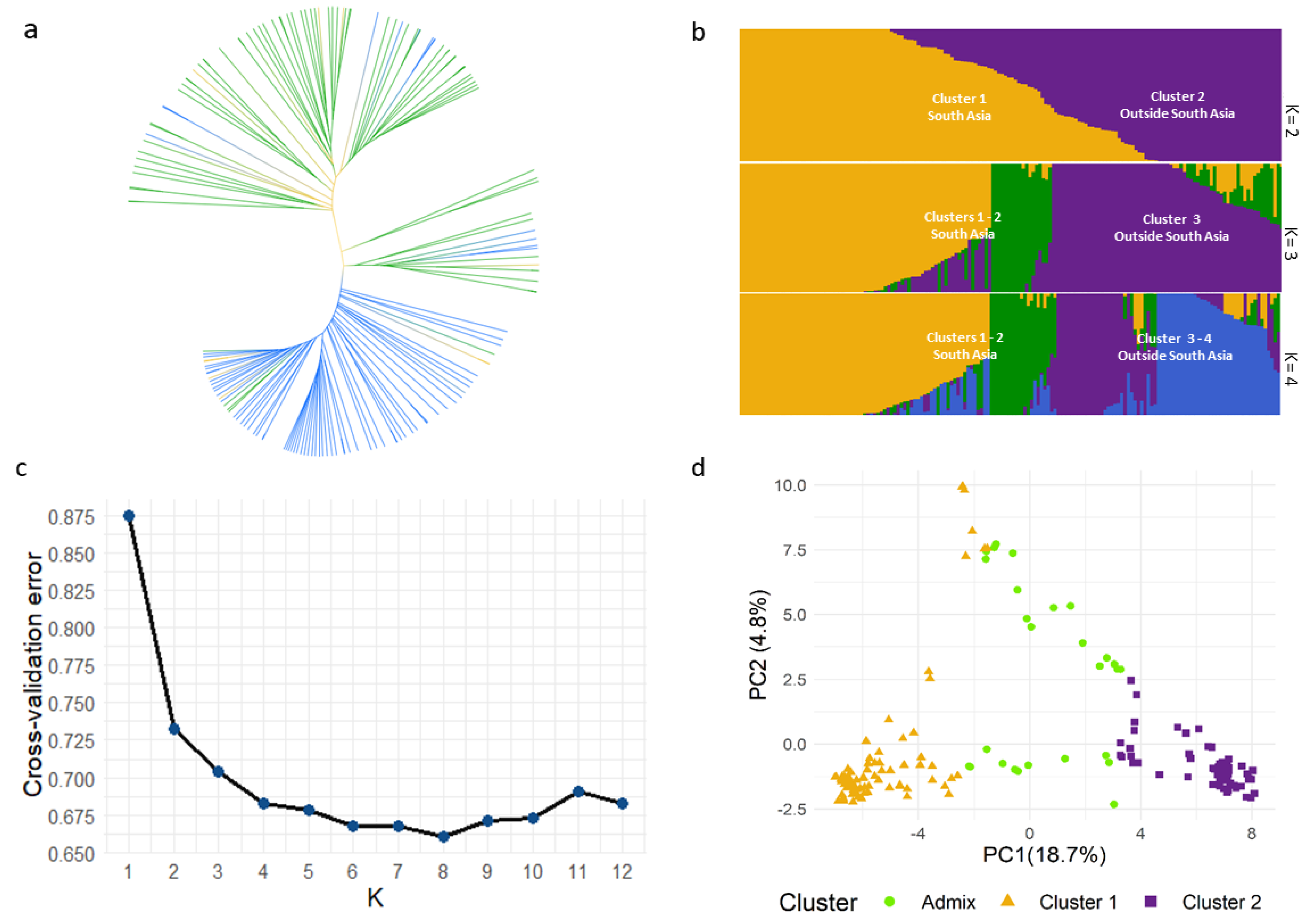
2.2. Cluster, Population Structure and Principal Component Analyses of B. juncea Diverse Panel
2.3. Variance Components, Basic Descriptive Statistics and Correlations between Total GSLs, Sinigrin and Gluconapin
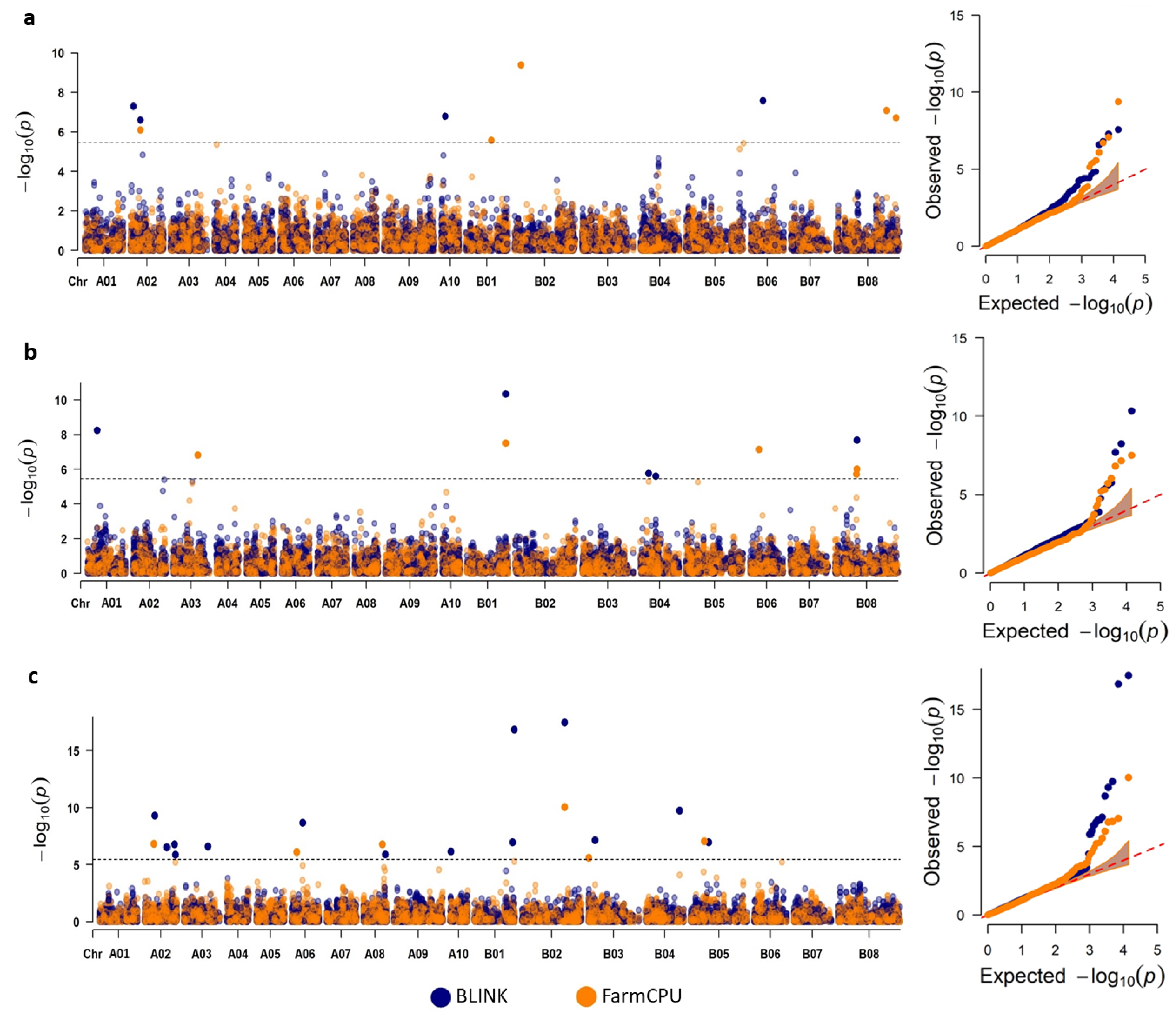
2.4. GWAS Using Multiple Models
2.5. Significant GWAS Hits Had Known and Potential GSL Genes in Their Vicinity
3. Discussion
3.1. Population Structure
3.2. Candidate Genes Identified in the Vicinity of Associated SNPs
4. Materials and Methods
4.1. Plant Materials and Growing Conditions
4.2. Glucosinolate Analysis
4.3. Bioinformatic Analyses and Data Processing
4.4. Statistical Analysis
4.5. Genome-Wide Association Analysis
4.6. Cluster, Population Structure and Principal Components Analysis
4.7. Candidate Genes within Significant SNPs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halkier, B.A.; Gershenzon, J. Biology and biochemistry of glucosinolates. Annu. Rev. Plant Biol. 2006, 57, 303–333. [Google Scholar] [CrossRef] [Green Version]
- Burow, M.; Halkier, B.A.; Kliebenstein, D.J. Regulatory networks of glucosinolates shape Arabidopsis thaliana fitness. Curr. Opin. Plant Biol. 2010, 13, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiari, M.; Rasmann, S. Genotypic variation in below-to aboveground systemic induction of glucosinolates mediates plant fitness consequences under herbivore attack. J. Chem. Ecol. 2019, 317–329. [Google Scholar] [CrossRef]
- Harun, S.; Abdullah-Zawawi, M.R.; Goh, H.H.; Mohamed-Hussein, Z.A. A comprehensive gene inventory for glucosinolate biosynthetic pathway in Arabidopsis thaliana. J. Agric. Food Chem. 2020, 68, 7281–7297. [Google Scholar] [CrossRef] [PubMed]
- Sønderby, I.E.; Geu-Flores, F.; Halkier, B.A. Biosynthesis of glucosinolates—Gene discovery and beyond. Trends Plant Sci. 2010, 15, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Augustine, R.; Mukhopadhyay, A.; Bisht, N.C. Targeted silencing of BjMYB28 transcription factor gene directs development of low glucosinolate lines in oilseed Brassica juncea. Plant Biotechnol. J. 2013, 11, 855–866. [Google Scholar] [CrossRef]
- Dixon, G.R. Vegetable brassicas and related crucifers: Origins and diversity of brassica and its relatives. Veg. Brassicas Relat. Crucif. 2006, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Raymer, P.L. Canola: An emerging oilseed crop. Trends New Crop. New Uses 2002, 1, 122–126. [Google Scholar]
- Love, H.K.; Rakow, G.; Raney, J.P.; Downey, R.K. Development of low glucosinolate mustard. Can. J. Plant Sci. 1990, 70, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Potts, D.A.; Rakow, G.W.; Males, D.R. Canola quality Brassica juncea, a new oilseed crop for the canadian prairies. In Proceedings of the 10th International Rapeseed Congress, Canberra, Australia, 26–29 September 1999. [Google Scholar]
- Norton, R.; Potter, T.; Haskins, B.; Mccaffery, D.; Bambach, R. Juncea Canola in the Low Rainfall Zones of Victoria and South Australia. Juncea Canola Growers Guide–Victoria and South Australia. Available online: http://anz.ipni.net/ipniweb/region/anz.nsf/0/CE50267DC5CD6D5385257AA10052C4E0/$FILE/ViCSAGrowersGuide.pdf (accessed on 26 September 2021).
- Woods, D.L.; Capcara, J.J.; Downey, R.K. The potential of mustard (Brassica juncea (L.) Coss) as an edible oil crop on the Canadian prairies. Can. J. Plant Sci. 1991, 71, 195–198. [Google Scholar] [CrossRef]
- Burton, W.; Pymer, S.; Salisbury, P.; Kirk, J.; Oram, R. Performance of Australian canola quality Brassica juncea breeding lines. In Proceedings of the 10th International Rapeseed Congress, Canberra, Australia, 26–29 September 1999; pp. 2–7. [Google Scholar]
- Merah, O. Genetic Variability in glucosinolates in seed of Brassica juncea: Interest in mustard condiment. J. Chem. 2015, 2015, 606142. [Google Scholar] [CrossRef] [Green Version]
- Misiewicz, I.; Skupińska, K.; Kowalska, E.; Lubiński, J.; Kasprzycka-Guttman, T. Sulforaphane-mediated induction of a phase 2 detoxifying enzyme NAD(P)H:Quinone reductase and apoptosis in human lymphoblastoid cells. Acta Biochim. Pol. 2004, 51, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M. Sulforaphane (SFN): An isothiocyanate in a cancer chemoprevention paradigm. Medicines 2015, 2, 141–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wang, J.; Li, Z.; Li, X.; He, Z.; Zhang, L.; Sha, T.; Lyu, X.; Chen, S.; Gu, Y.; et al. Genomic signatures of vegetable and oilseed allopolyploid Brassica juncea and genetic loci controlling the accumulation of glucosinolates. Plant Biotechnol. J. 2021, 19, 2619–2628. [Google Scholar] [CrossRef]
- Sharma, M.; Mukhopadhyay, A.; Gupta, V.; Pental, D.; Pradhan, A.K. BjuB.CYP79F1 regulates synthesis of propyl fraction of aliphatic glucosinolates in oilseed mustard Brassica juncea: Functional validation through genetic and transgenic approaches. PLoS ONE 2016, 11, e0150060. [Google Scholar] [CrossRef]
- Moose, S.P.; Mumm, R.H. Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol. 2008, 147, 969–977. [Google Scholar] [CrossRef] [Green Version]
- Harper, A.L.; He, Z.; Langer, S.; Havlickova, L.; Wang, L.; Fellgett, A.; Gupta, V.; Kumar Pradhan, A.; Bancroft, I. Validation of an associative transcriptomics platform in the polyploid crop species Brassica juncea by dissection of the genetic architecture of agronomic and quality traits. Plant J. 2020, 103, 1885–1893. [Google Scholar] [CrossRef]
- Rout, K.; Sharma, M.; Gupta, V.; Mukhopadhyay, A.; Sodhi, Y.S.; Pental, D.; Pradhan, A.K. Deciphering allelic variations for seed glucosinolate traits in oilseed mustard (Brassica juncea) using two bi-parental mapping populations. Theor. Appl. Genet. 2015, 128, 657–666. [Google Scholar] [CrossRef]
- Rout, K.; Yadav, B.G.; Yadava, S.K.; Mukhopadhyay, A.; Gupta, V.; Pental, D.; Pradhan, A.K. QTL landscape for oil content in Brassica juncea: Analysis in multiple bi-parental populations in high and “0” erucic background. Front. Plant Sci. 2018, 9, 871. [Google Scholar] [CrossRef] [Green Version]
- Ramchiary, N.; Bisht, N.C.; Gupta, V.; Mukhopadhyay, A.; Arumugam, N.; Sodhi, Y.S.; Pental, D.; Pradhan, A.K. qtl analysis reveals context-dependent loci for seed glucosinolate trait in the oilseed Brassica juncea: Importance of recurrent selection backcross scheme for the identification of “true” QTL. Theor. Appl. Genet. 2007, 116, 77–85. [Google Scholar] [CrossRef]
- Raman, H.; Raman, R.; Kilian, A.; Detering, F.; Carling, J.; Coombes, N.; Diffey, S.; Kadkol, G.; Edwards, D.; McCully, M.; et al. Genome-wide delineation of natural variation for pod shatter resistance in Brassica napus. PLoS ONE 2014, 9, e101673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Huang, H.; Yi, X.; Zhang, Y.; Yang, Q.; Zhang, C.; Fan, C.; Zhou, Y. Dissection of genetic architecture for glucosinolate accumulations in leaves and seeds of Brassica napus by genome-wide association study. Plant Biotechnol. J. 2020, 18, 1472–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, C.; Jia, L.; Fu, F.; Zhao, H.; Lu, K.; Wei, L.; Xu, X.; Liang, Y.; Li, S.; Wang, R.; et al. Genome-wide association mapping and identification of candidate genes for fatty acid composition in Brassica napus L. using SNP Markers. BMC Genom. 2017, 18, 232. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wu, Z.; Li, Z.; Zhang, Q.; Hu, J.; Xiao, Y.; Cai, D.; Wu, J.; King, G.J.; Li, H.; et al. Dissection of the genetic architecture of three seed-quality traits and consequences for breeding in Brassica napus. Plant Biotechnol. J. 2018, 16, 1336–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Akhatar, J.; Kaur, H.; Atri, C.; Mittal, M.; Goyal, A.; Pant, U.; Kaur, G.; Banga, S.S. Genome-wide association mapping for key seed metabolites using a large panel of natural and derived forms of Brassica rapa L. Ind. Crops Prod. 2021, 159, 113073. [Google Scholar] [CrossRef]
- Sudan, J.; Singh, R.; Sharma, S.; Salgotra, R.K.; Sharma, V.; Singh, G.; Sharma, I.; Sharma, S.; Gupta, S.K.; Zargar, S.M. DdRAD sequencing-based identification of inter-genepool SNPS and association analysis in Brassica juncea. BMC Plant Biol. 2019, 19, 594. [Google Scholar] [CrossRef] [Green Version]
- Paritosh, K.; Gupta, V.; Yadava, S.K.; Singh, P.; Pradhan, A.K.; Pental, D. RNA-Seq based snps for mapping in Brassica juncea (AABB): Synteny analysis between the two constituent genomes A (from B. rapa) and B (from B. nigra) shows highly divergent gene block arrangement and unique block fragmentation patterns. BMC Genom. 2014, 15, 396. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, C.; Zhao, N.; Zhang, L.; Hu, Z.; Chen, S.; Zhang, M. Chinese root-type mustard provides phylogenomic insights into the evolution of the multi-use diversified allopolyploid Brassica juncea. Mol. Plant 2018, 11, 512–514. [Google Scholar] [CrossRef] [Green Version]
- Akhatar, J.; Singh, M.P.; Sharma, A.; Kaur, H.; Kaur, N.; Sharma, S.; Bharti, B.; Sardana, V.K.; Banga, S.S. Association mapping of seed quality traits under varying conditions of nitrogen application in Brassica juncea L. Czern & Coss. Front. Genet. 2020, 11, 744. [Google Scholar] [CrossRef]
- Hirai, M.Y.; Klein, M.; Fujikawa, Y.; Yano, M.; Goodenowe, D.B.; Yamazaki, Y.; Kanaya, S.; Nakamura, Y.; Kitayama, M.; Suzuki, H.; et al. Elucidation of gene-to-gene and metabolite-to-gene networks in Arabidopsis by integration of metabolomics and transcriptomics. J. Biol. Chem. 2005, 280, 25590–25595. [Google Scholar] [CrossRef] [Green Version]
- Hirai, M.Y.; Sugiyama, K.; Sawada, Y.; Tohge, T.; Obayashi, T.; Suzuki, A.; Araki, R.; Sakurai, N.; Suzuki, H.; Aoki, K.; et al. Omics-based identification of Arabidopsis Myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6478–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wentzell, A.M.; Rowe, H.C.; Hansen, B.G.; Ticconi, C.; Halkier, B.A.; Kliebenstein, D.J. Linking metabolic QTLs with network and cis-eqtls controlling biosynthetic pathways. PLoS Genet. 2007, 3, 1687–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kaminaga, Y.; Cooper, B.; Pichersky, E.; Dudareva, N.; Chapple, C. Benzoylation and sinapoylation of glucosinolate R-groups in Arabidopsis. Plant J. 2012, 72, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skirycz, A.; Reichelt, M.; Burow, M.; Birkemeyer, C.; Rolcik, J.; Kopka, J.; Zanor, M.I.; Gershenzon, J.; Strnad, M.; Szopa, J.; et al. DOF transcription factor AtDof1.1 (OBP2) is part of a regulatory network controlling glucosinolate biosynthesis in Arabidopsis. Plant J. 2006, 47, 10–24. [Google Scholar] [CrossRef]
- Gigolashvili, T.; Yatusevich, R.; Berger, B.; Müller, C.; Flügge, U.I. The R2R3-MYB transcription factor HAG1/MYB28 is a regulator of methionine-derived glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 2007, 51, 247–261. [Google Scholar] [CrossRef]
- Sønderby, I.E.; Hansen, B.G.; Bjarnholt, N.; Ticconi, C.; Halkier, B.A.; Kliebenstein, D.J. A systems biology approach identifies a R2R3 MYB gene subfamily with distinct and overlapping functions in regulation of aliphatic glucosinolates. PLoS ONE 2007, 2, e1322. [Google Scholar] [CrossRef]
- Celenza, J.L.; Quiel, J.A.; Smolen, G.A.; Merrikh, H.; Silvestro, A.R.; Normanly, J.; Bender, J. The Arabidopsis ATR1 Myb transcription factor controls indolic glucosinolate homeostasis. Plant Physiol. 2005, 137, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Frerigmann, H.; Gigolashvili, T. Update on the role of R2R3-MYBs in the regulation of glucosinolates upon sulfur deficiency. Front. Plant Sci. 2014, 5, 626. [Google Scholar] [CrossRef] [Green Version]
- Kroymann, J.; Textor, S.; Tokuhisa, J.G.; Falk, K.L.; Bartram, S.; Gershenzon, J.; Mitchell-Olds, T. A gene controlling variation in Arabidopsis glucosinolate composition is part of the methionine chain elongation pathway. Plant Physiol. 2001, 127, 1077–1088. [Google Scholar] [CrossRef]
- Benderoth, M.; Pfalz, M.; Kroymann, J. Methylthioalkylmalate synthases: Genetics, ecology and evolution. Phytochem. Rev. 2009, 8, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hansen, B.G.; Ober, J.A.; Kliebenstein, D.J.; Halkier, B.A. Subclade of flavin-monooxygenases involved in aliphatic glucosinolate biosynthesis. Plant Physiol. 2008, 148, 1721–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Kristiansen, K.A.; Hansen, B.G.; Halkier, B.A. Cellular and subcellular localization of flavin-monooxygenases involved in glucosinolate biosynthesis. J. Exp. Bot. 2011, 62, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Huseby, S.; Koprivova, A.; Lee, B.R.; Saha, S.; Mithen, R.; Wold, A.B.; Bengtsson, G.B.; Kopriva, S. Diurnal and light regulation of sulphur assimilation and glucosinolate biosynthesis in Arabidopsis. J. Exp. Bot. 2013, 64, 1039–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sul, J.H.; Martin, L.S.; Eskin, E. Population structure in genetic studies: Confounding factors and mixed models. PLoS Genet. 2018, 14, e1007309. [Google Scholar] [CrossRef] [PubMed]
- Akhatar, J.; Goyal, A.; Kaur, N.; Atri, C.; Mittal, M.; Singh, M.P.; Kaur, R.; Rialch, I.; Banga, S.S. Genome-wide association analyses to understand genetic basis of flowering and plant height under three levels of nitrogen application in Brassica juncea (L.) Czern & Coss. Sci. Rep. 2021, 11, 4278. [Google Scholar] [CrossRef]
- Vaughan, J.G.; Gordon, E.I. A taxonomic study of Brassica juncea using the techniques of electrophoresis, gas-liquid chromatography and serology. Ann. Bot. 1973, 37, 167–184. [Google Scholar] [CrossRef]
- Gland, A.; Röbbelen, G.; Thies, W. Variation of alkenyl glucosinolates in seeds of Brassica species. Z. Pflanzenzüchtg 1981, 87, 96–110. [Google Scholar]
- Sodhi, Y.S.; Mukhopadhyay, A.; Arumugam, N.; Verma, J.K.; Gupta, V.; Pental, D.; Pradhan, A.K. Genetic analysis of total glucosinolate in crosses involving a high glucosinolate indian variety and a low glucosinolate line of Brassica Juncea. Plant Breed. 2002, 121, 508–511. [Google Scholar] [CrossRef]
- Velasco, L.; Becker, H.C. Variability for seed glucosinolates in a germplasm collection of the genus brassica. Genet. Resour. Crop Evol. 2000, 47, 231–238. [Google Scholar] [CrossRef]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Kaler, A.S.; Gillman, J.D.; Beissinger, T.; Purcell, L.C. Comparing different statistical models and multiple testing corrections for association mapping in soybean and maize. Front. Plant Sci. 2020, 10, 1794. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.; Li, J.; Hu, W.; Yu, J.; Khan, S.U.; Khan, M.H.U.; Xie, G.; Wang, J.; Wang, L. Uncovering genomic regions controlling plant architectural traits in hexaploid wheat using different GWAS models. Sci. Rep. 2021, 11, 6767. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Liu, S.; Meng, X.; Sun, T.; Deng, Y.; Kong, W.; Peng, Z.; Li, Y. Correction to: Uncovering the genetic mechanisms regulating panicle architecture in rice with GPWAS and GWAS. BMC Genom. 2021, 22, 86. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience 2018, 8, giy154. [Google Scholar] [CrossRef]
- Harun, S.; Abdullah-Zawawi, M.R.; A-Rahman, M.R.A.; Muhammad, N.A.N.; Mohamed-Hussein, Z.A. SuCComBase: A manually curated repository of plant sulfur-containing compounds. Database 2019, 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.G.; Singh, K.B. Characterization of salicylic acid-responsive, Arabidopsis Dof domain proteins: Overexpression of OBP3 leads to growth defects. Plant J. 2000, 21, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Kliebenstein, D.J.; Lambrix, V.M.; Reichelt, M.; Gershenzon, J.; Mitchell-Olds, T. Gene duplication in the diversification of secondary metabolism: Tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell 2001, 13, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Burow, M.; Atwell, S.; Francisco, M.; Kerwin, R.E.; Halkier, B.A.; Kliebenstein, D.J. The glucosinolate biosynthetic gene AOP2 mediates feed-back regulation of jasmonic acid signaling in Arabidopsis. Mol. Plant 2015, 8, 1201–1212. [Google Scholar] [CrossRef] [Green Version]
- Benstein, R.M.; Ludewig, K.; Wulfert, S.; Wittek, S.; Gigolashvili, T.; Frerigmann, H.; Gierth, M.; Flügge, U.I.; Krueger, S. Arabidopsis phosphoglycerate dehydrogenase1 of the phosphoserine pathway is essential for development and required for ammonium assimilation and tryptophan biosynthesis. Plant Cell 2013, 25, 5011–5029. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Tohge, T.; Fernie, A.R.; Hoefgen, R. The effect of single and multiple serat mutants on serine and sulfur metabolism. Front. Plant Sci. 2018, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Niemiro, A.; Cysewski, D.; Brzywczy, J.; Wawrzyńska, A.; Sieńko, M.; Poznański, J.; Sirko, A. Similar but not identical—Binding properties of LSU (Response to Low Sulfur) proteins from Arabidopsis Thaliana. Front. Plant Sci. 2020, 11, 1246. [Google Scholar] [CrossRef] [PubMed]
- Aarabi, F.; Naake, T.; Fernie, A.R.; Hoefgen, R. Coordinating sulfur pools under sulfate deprivation. Trends Plant Sci. 2020, 25, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, Y.; Tanaka, M.; Tabata, R.; Ogawa-Ohnishi, M.; Matsubayashi, Y. Shoot-to-Root Mobile Polypeptides Involved in Systemic Regulation of Nitrogen Acquisition. Nat. Plants 2017, 3, 17029. [Google Scholar] [CrossRef]
- Grubb, C.D.; Zipp, B.J.; Ludwig-Müller, J.; Masuno, M.N.; Molinski, T.F.; Abel, S. Arabidopsis glucosyltransferase UGT74B1 functions in glucosinolate biosynthesis and auxin homeostasis. Plant J. 2004, 40, 893–908. [Google Scholar] [CrossRef]
- Grubb, C.D.; Zipp, B.J.; Kopycki, J.; Schubert, M.; Quint, M.; Lim, E.K.; Bowles, D.J.; Pedras, M.S.C.; Abel, S. Comparative analysis of Arabidopsis UGT74 glucosyltransferases reveals a special role of UGT74C1 in glucosinolate biosynthesis. Plant J. 2014, 79, 92–105. [Google Scholar] [CrossRef]
- Romero, L.C.; Aroca, M.Á.; Laureano-Marín, A.M.; Moreno, I.; García, I.; Gotor, C. Cysteine and cysteine-related signaling pathways in Arabidopsis thaliana. Mol. Plant 2014, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Aarabi, F.; Kusajima, M.; Tohge, T.; Konishi, T.; Gigolashvili, T.; Takamune, M.; Sasazaki, Y.; Watanabe, M.; Nakashita, H.; Fernie, A.R.; et al. Sulfur deficiency-induced repressor proteins optimize glucosinolate biosynthesis in plants. Sci. Adv. 2016, 2, e1601087. [Google Scholar] [CrossRef] [Green Version]
- Borpatragohain, P.; Rose, T.J.; Liu, L.; Barkla, B.J.; Raymond, C.A.; King, G.J. Remobilization and fate of sulphur in mustard. Ann. Bot. 2019, 124, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Murray, K.D.; Borevitz, J.O. Axe: Rapid, Competitive sequence read demultiplexing using a trie. Bioinformatics 2018, 34, 3924–3925. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassambara, A. Ggpubr: ‘Ggplot2′ Based Publication Ready Plots 2020. 2021. Available online: https://github.com/kassambara/ggpubr (accessed on 1 September 2021).
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckler, E.; Zhang, Z. User Manual for Genomic Association and Prediction Integrated Tool (GAPIT). 2018. Version 3. Available online: https://zzlab.net/GAPIT/gapit_help_document.pdf (accessed on 27 August 2021).
- Segura, V.; Vilhjálmsson, B.J.; Platt, A.; Korte, A.; Seren, Ü.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Tian, F.; Pan, Y.; Buckler, E.S.; Zhang, Z. A SUPER powerful method for genome wide association study. PLoS ONE 2014, 9, e0107684. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. RMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinform. 2021. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Niu, S.; Song, Q.; Koiwa, H.; Qiao, D.; Zhao, D.; Chen, Z.; Liu, X.; Wen, X. Genetic diversity, linkage disequilibrium, and population structure analysis of the tea plant (Camellia sinensis) from an origin center, guizhou plateau, using genome-wide SNPs developed by genotyping-by-sequencing. BMC Plant Biol. 2019, 19, 328. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.M. Pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; pp. 1–69. ISBN 978-0-387-98141-3. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cock, P.J.A.; Chilton, J.M.; Grüning, B.; Johnson, J.E.; Soranzo, N. NCBI BLAST+ Integrated into Galaxy. Gigascience 2015, 4, s13742-015-0080-7. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Trait a | Peak SNP | p-Value | PVE b | Model c | Candidate Gene | Homologous Gene in Arabidopsis | % Amino Acid Identity | Distance to Peak SNP [kb] | Arabidopsis ID | Gene Description |
|---|---|---|---|---|---|---|---|---|---|---|
| TGSL | A02_3567961 | 5.08 × 10−8 | 6.63 | B | BjuA041358 | GSTF11 [33,34,35] | 69.16 | −39.61 | AT3G03190.1 | GSL core structure synthesis |
| BjuA041338 | SCPL17 [36] | 61.43 | 68.54 | AT3G12203.3 | GSL side-chain modification | |||||
| A02_11235033 | 2.54 × 10−7 | 5.83 | B, F, M, S | BjuA045411 | OBP2 [37] | 84.92 | −128.81 | AT1G07640.1 | GSL regulation | |
| B02_7295738 | 4.09 × 10−10 | 11.41 | F, S | BjuB047551 | AAP4 | 94.21 | 213.38 | AT5G63850.1 | potential GSL gene | |
| BjuB047557 | SAL1 | 64.76 | 246.34 | AT5G63980.1 | potential GSL gene | |||||
| B08_66155255 | 1.90 × 10−7 | 37.03 | F | BjuB019211 | CYP18-3 | 65.48 | −0.7 | AT4G38740.1 | potential GSL gene | |
| BjuB019215 | Probable 2-ODDd | 61.77 | −17.56 | AT5G05600.1 | potential GSL gene | |||||
| SIN | A03_27702263 | 1.50 × 10−7 | 3.76 | F, S | BjuA042263 | MYB28 [38,39] | 79.95 | −118.32 | AT5G61420.2 | GSL regulation |
| BjuA042229 | MYB34 [40,41] | 71.32 | 115.48 | AT5G60890.1 | GSL regulation | |||||
| BjuA042223 | MAM1 [42,43] | 82.72 | 160.65 | AT5G23010.3 | GSL side-chain elongation | |||||
| B04_9016612 | 5.04 × 10−6 | 6.84 | B | BjuB028146 | FMOGS-OX5 [44,45] | 69.41 | −1.51 | AT1G12140.3 | GSL side-chain modification | |
| B04_17138489 | 2.51 × 10−6 | 11.71 | B | BjuB028703 | PSAT1 | 83.57 | 12.75 | AT4G35630.1 | potential GSL gene | |
| GNP | A02_34185026 | 1.64 × 10−7 | 11.24 | B | BjuA033112 | LSU2 | 86.022 | 5.75 | AT5G24660.1 | potential GSL gene |
| A02_34995417 | 1.29 × 10−6 | 0.72 | B | BjuA002140 | MYB28 [38,39] | 67.46 | 81.62 | AT5G61420.1 | GSL regulation | |
| BjuA001524 | MYB34 [40,41] | 72.00 | 96.36 | AT5G60890.1 | GSL regulation | |||||
| A10_999168 | 6.85 × 10−7 | 10.72 | B | BjuA037371 | GRXS11 | 96.97 | −105.45 | AT1G06830.1 | potential GSL gene | |
| BjuA037341 | UGT71C3 | 79.19 | 115.13 | AT1G07260.1 | potential GSL gene | |||||
| B01_44925254 | 1.38 × 10−17 | 7.15 | B, S | BjuB006588 | RER3 | 78.44 | −105.02 | AT3G08640.1 | potential GSL gene | |
| BjuB006607 | CYSD1 | 73.27 | −213.34 | AT3G04940.2 | potential GSL gene | |||||
| B02_48309648-753 | 3.35 × 10−18 | 2.80 | B, F | BjuB009816 | HY5 [46] | 89.94 | 180.71 | AT5G11260.1 | GSL regulation | |
| B03_474869 | 2.49 × 10−6 | 6.03 | F | BjuB005751 | SDI2 | 82.50 | 23.76 | AT1G04770.1 | potential GSL gene | |
| B03_7408562 | 7.07 × 10−8 | 4.78 | B, M | BjuB003011 | ALDH2B7 | 91.01 | 135.05 | AT1G23800.1 | potential GSL gene |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tandayu, E.; Borpatragohain, P.; Mauleon, R.; Kretzschmar, T. Genome-Wide Association Reveals Trait Loci for Seed Glucosinolate Accumulation in Indian Mustard (Brassica juncea L.). Plants 2022, 11, 364. https://doi.org/10.3390/plants11030364
Tandayu E, Borpatragohain P, Mauleon R, Kretzschmar T. Genome-Wide Association Reveals Trait Loci for Seed Glucosinolate Accumulation in Indian Mustard (Brassica juncea L.). Plants. 2022; 11(3):364. https://doi.org/10.3390/plants11030364
Chicago/Turabian StyleTandayu, Erwin, Priyakshee Borpatragohain, Ramil Mauleon, and Tobias Kretzschmar. 2022. "Genome-Wide Association Reveals Trait Loci for Seed Glucosinolate Accumulation in Indian Mustard (Brassica juncea L.)" Plants 11, no. 3: 364. https://doi.org/10.3390/plants11030364
APA StyleTandayu, E., Borpatragohain, P., Mauleon, R., & Kretzschmar, T. (2022). Genome-Wide Association Reveals Trait Loci for Seed Glucosinolate Accumulation in Indian Mustard (Brassica juncea L.). Plants, 11(3), 364. https://doi.org/10.3390/plants11030364