1. Introduction
Wuzhimaotao (
Ficus hirta Vahl) is a common Chinese herbal medicine in the Lingnan area of China. It is a folk homologous plant of food and medicine, which is called Guangdong ginseng. It is used by Hakka people in southern China to treat diseases such as spleen deficiency, tuberculosis, weakness, rheumatism, night sweats, and agalactism [
1,
2,
3]. Wuzhimaotao uses the roots of
Ficus hirta Vahl as medicine. It is mainly distributed in Guangdong, Guangxi, Jiangxi, Fujian, Yunnan, Hong Kong of China, and Southeast Asian countries. The isolated and identified compounds in Wuzhimaotao mainly include coumarins, flavonoids, and volatile oil. Modern pharmacological studies show that these compounds have antioxidant, anti-inflammatory, antibacterial, antiviral, and antitumor effects [
4].
In fact, the main pharmacodynamic components of Chinese herbal medicine are usually plant secondary metabolites. Studies shows that stress plays an important role in the formation of genuine medicinal materials [
5]. When plants are infected by biological or abiotic factors, they can synthesize and accumulate a series of low relative molecular weight compounds with disease resistance through the expression of resistance genes in vivo to resist the invasion of pathogens. These substances are commonly known as phytoalexins. Phytoalexins are usually secondary metabolites, which are a large class of small molecular compounds produced by plants in the process of secondary metabolism [
6]. Common secondary metabolites include alkaloids, flavonoids, terpenoids, anthraquinones, coumarins, and lignins. Stress conditions can promote the accumulation of plant secondary metabolites, and the accumulation of secondary metabolites can improve the stress resistance of plants. Therefore, the measures to improve plant stress resistance are expected to promote the accumulation of plant secondary metabolites in an effort to improve the genuine quality of traditional Chinese medicine [
5]. The active components (coumarins, flavonoids, and volatile oil) of Wuzhimaotao are common plant secondary metabolites. The quality of the traditional Chinese medicine Wuzhimaotao and the content of its active components are expected to improve as stress conditions or other methods are used to improve its stress resistance.
We found that hydrogen is involved in plant stress responses by regulating plant hormone signal transduction [
7]. Hydrogen water treatment can improve the disease resistance of plants and the salt and drought resistance of rice seedlings. Stress treatment can also improve the ability of rice to release hydrogen [
7]. Hydrogen water treatment can reduce the harm of heavy metal stress to plants [
8,
9,
10]. We also found that hydrogen water can promote the growth of mung bean and rice roots and seedlings [
7]. Therefore, we put forward the concept of hydrogen agriculture, i.e., using hydrogen water and other kinds of hydrogen fertilizer to promote the growth of crops and improve their stress resistance, in order to reduce the use of pesticides and chemical fertilizers, protect the ecological environment, and ensure food safety [
11]. In addition, hydrogen fertilizers could be applied to the cultivation of traditional Chinese medicine to improve the content of effective components, yield, stress resistance, and, therefore, the quality of genuine medicinal materials.
In order to determine whether hydrogen molecules can increase the content of plant secondary metabolites and the molecular mechanism of hydrogen regulating the synthesis of secondary metabolites, the metabolome and transcriptome of Wuzhimaotao were analyzed.
3. Discussion
Although the biological effects of hydrogen have long been noticed in botany and medicine [
12,
13,
14], they attracted extensive attention only after 2007 [
15]. Presently, it is understood that the mechanism of the hydrogen biological effect is that hydrogen has both antioxidant effects and signal molecule effects [
16,
17]. Our previous study found that hydrogen affects plant growth, development, and stress adaptation by participating in the regulation of plant hormone signaling pathways [
7,
18]. Because the improvement of plant stress resistance is often manifested in the accumulation of secondary metabolites [
19,
20,
21], cultivation using hydrogen water may improve the accumulation of secondary metabolites, which may be conducive to improving the quality of traditional Chinese medicine.
Wuzhimaotao is a famous traditional herbal medicine with homology of medicine and food in southern China that has been used for centuries. It is the dry root of
Ficus hirta, a mulberry plant. The antioxidant, anti-inflammatory, antibacterial, antiviral, and antitumor effects of Wuzhimaotao have attracted more and more attention. It was reported that the main active components of Wuzhimaotao include coumarins, such as isopsoralen lactone, bergamot lactone, and psoralen, as well as flavonoids, such as apigenin and hesperidin [
4]. It was reported that 70 compounds were isolated in Wuzhimaotao, of which 30 phenylpropanoid compounds were the most abundant [
22]. The phenylpropane metabolic pathway in plants is an important pathway of secondary metabolism. All substances containing a phenylpropane skeleton are direct or indirect products of this pathway [
23]. The phenylpropanoid biosynthetic pathway is activated under stress conditions, resulting in accumulation of various phenolic compounds which, among other roles, have the potential to scavenge harmful reactive oxygen species (ROS) [
24,
25,
26]. In recent years, metabolomics integrated with transcriptomics has been widely used to investigate the biosynthesis of metabolites in plants [
27,
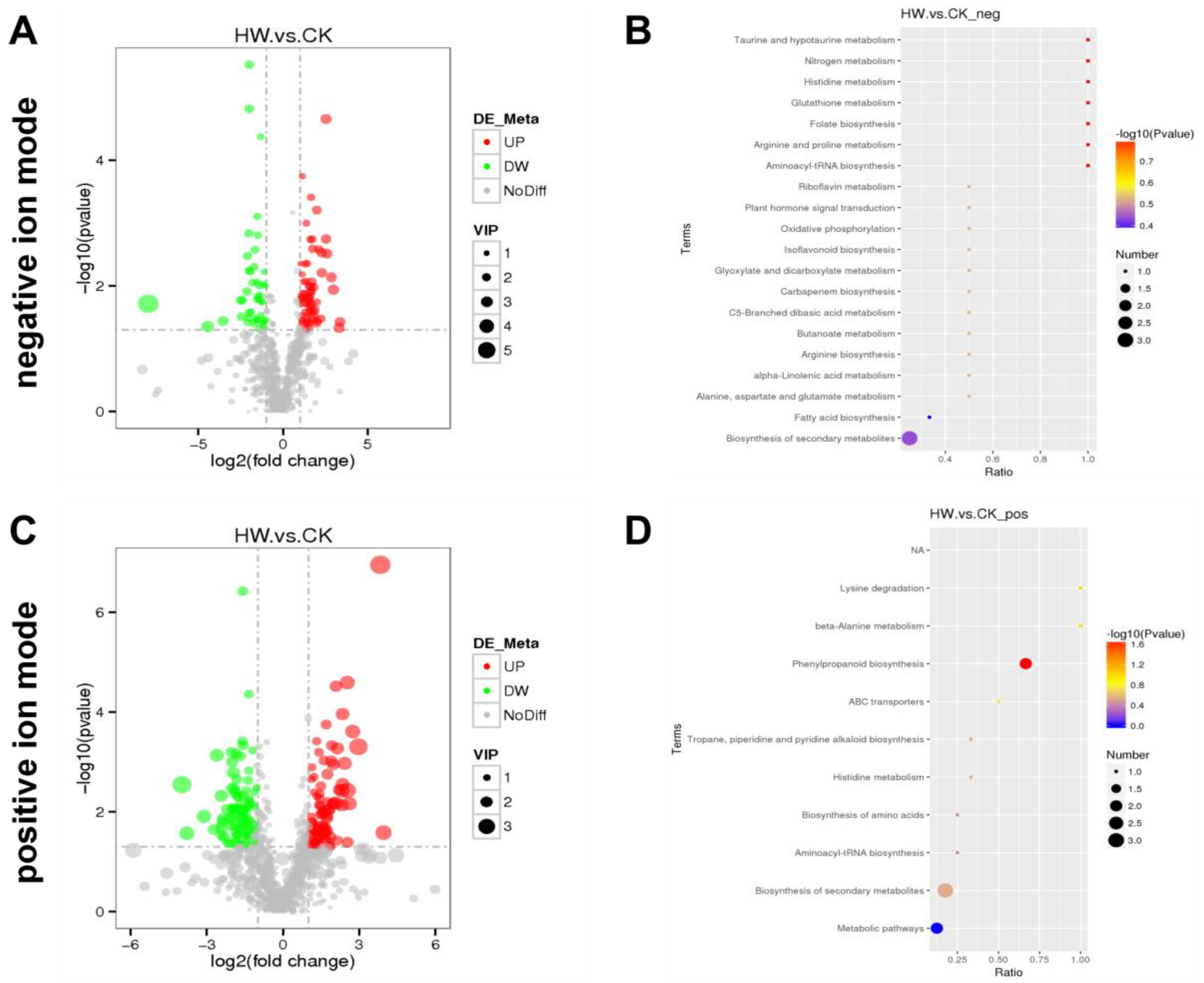
28]. In this study, hydrogen water was used to treat the famous Chinese southern medicine Wuzhimaotao, and then the metabolome and transcriptome were analyzed. Metabolomic analysis showed that there were a total of 277 significantly different metabolites between the hydrogen-water-treated group and the control group, including 148 upregulated and 129 downregulated metabolites. Among the upregulated metabolites, naringin (ID: Com_788_neg; fold change: 3.05), bergaptol (ID: Com_1406_neg; fold change: 2.28), hesperidin (ID: Com_3438_pos; fold change: 2.25), and a benzofuran (ID: Com_1053_neg; fold change: 2.19) were found (
Table S1), all of which are phenylpropane compounds. As their congeners and derivatives, narigenin, bergapten, hesperidin, and benzofuran compounds of were isolated and identified as the active ingredients in Wuzhimaotao. Psoralen, a benzofuran derivative, is one of the most important active components of Wuzhimaotao. Recently, many benzofuran compounds were isolated from Wuzhimaotao, such as 3-[6-(5-O-β-D-glucopyranosyl) benzofuranyl] methyl propionate, (E)-3-[5-(6-hydroxy) benzofuranyl] propenoic acid, (E)-3-[5-(6-methoxy) benzofuranyl] propenoic acid, (Z)-3-[5-(6-O-β-D-glucopyranosyl) benzofuranyl] methyl propenoate, and S-3-[2,3-dihydro-6-hydroxy-2-(1-hydroxy-1-methylethyl)-5-benzofuranyl] methyl propionate [
22]. Bergapten, psoralen, and other benzofurans belong to coumarins, and naringin and hesperidin belong to flavonoids. Coumarins and flavonoids are formed through the phenylpropanoid biosynthesis pathway. The results of the pathway enrichment analysis show that the significantly different metabolites were mainly associated with phenylpropanoid biosynthesis. The results show that hydrogen water treatment promoted the accumulation of active components in Wuzhimaotao, many of which were produced by phenylpropane synthesis. Therefore, planting Wuzhimaotao with hydrogen water may help to improve the content of the active components of Wuzhimaotao and improve the quality of traditional Chinese medicine.
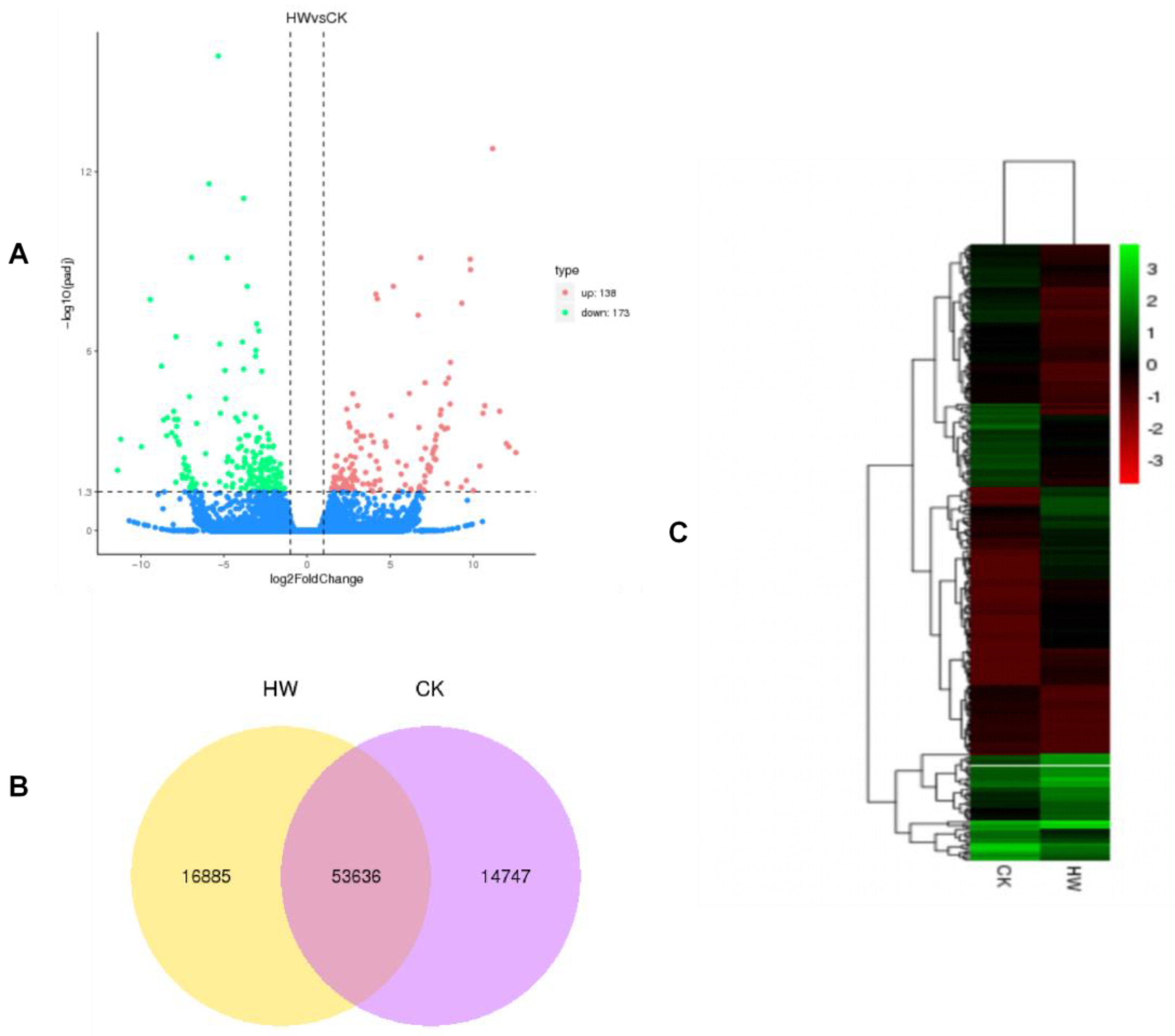
Transcriptome analysis showed that there were 311 significantly different DEGs between the hydrogen water treatment group and the control group, including 138 upregulated and 173 downregulated unigenes. KEGG pathway analysis found that transcripts regulated in the hydrogen-water-treated
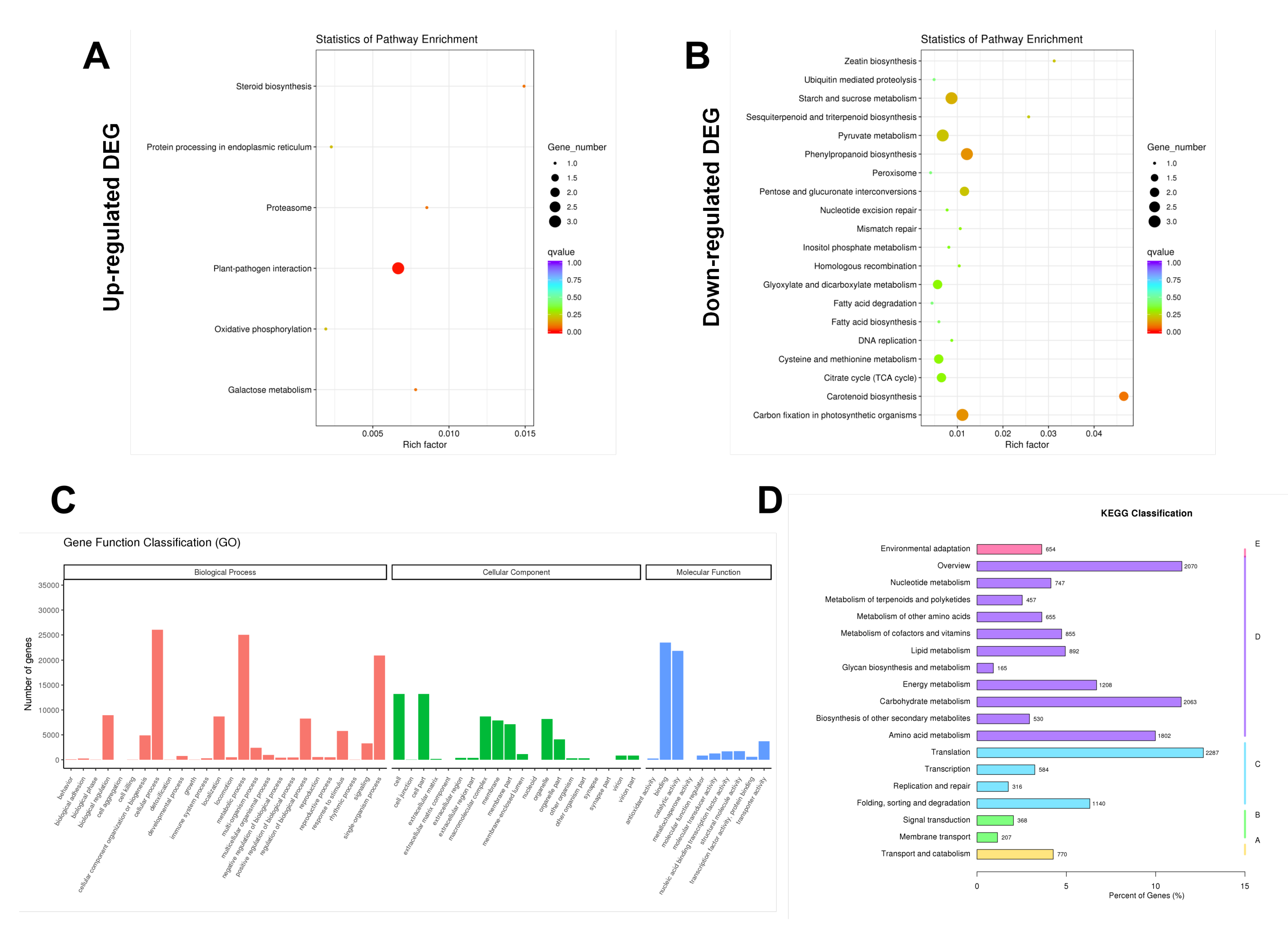
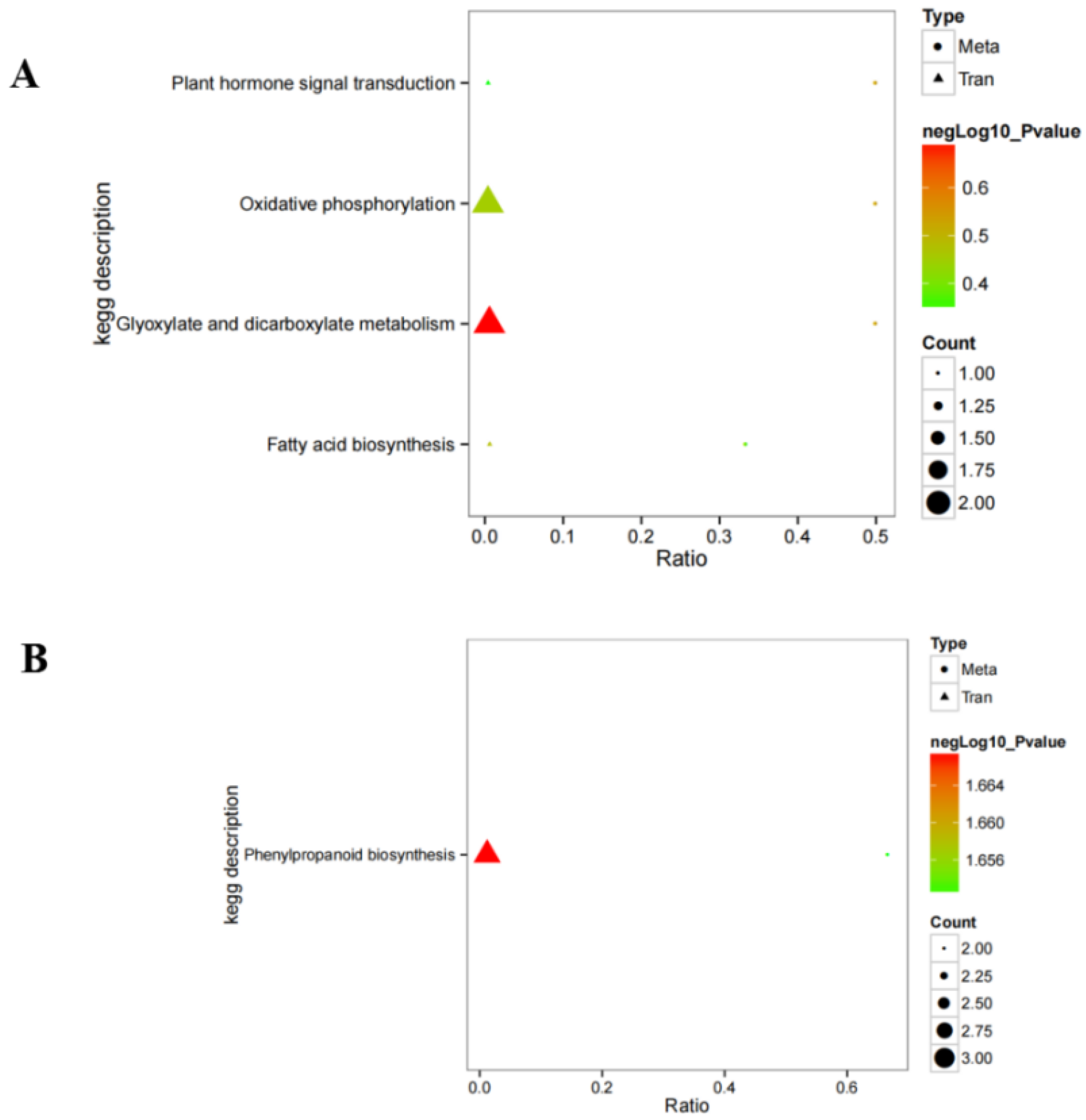
Ficus hirta Vahl roots could be mapped to signaling pathways, such as carotenoid biosynthesis, phenylpropanoid biosynthesis, and plant-pathogen interaction. The results of combined analysis of the metabolome and transcriptome showed that there were five enriched pathways: phenylpropanoid biosynthesis, plant hormone signal transduction, oxidative phosphorylation, glyoxylate and dicarboxylate metabolism, and fatty acid biosynthesis. The results show that hydrogen water treatment affected the phenylpropanoid biosynthesis and plant hormone signal transduction in
Ficus hirta Vahl roots.
Figure 4B shows that the DEGs of the phenylpropane synthesis pathway are downregulated, which seems to be inconsistent with the upregulation of coumarin and flavonoid metabolites shown by metabolome analysis. Comparing the KEGG enrichment obtained from metabolome and transcriptome analysis, we found that the phenylpropane synthesis pathway of the metabolome (
Figure S1) is different from that of the transcriptome (
Figure S2).
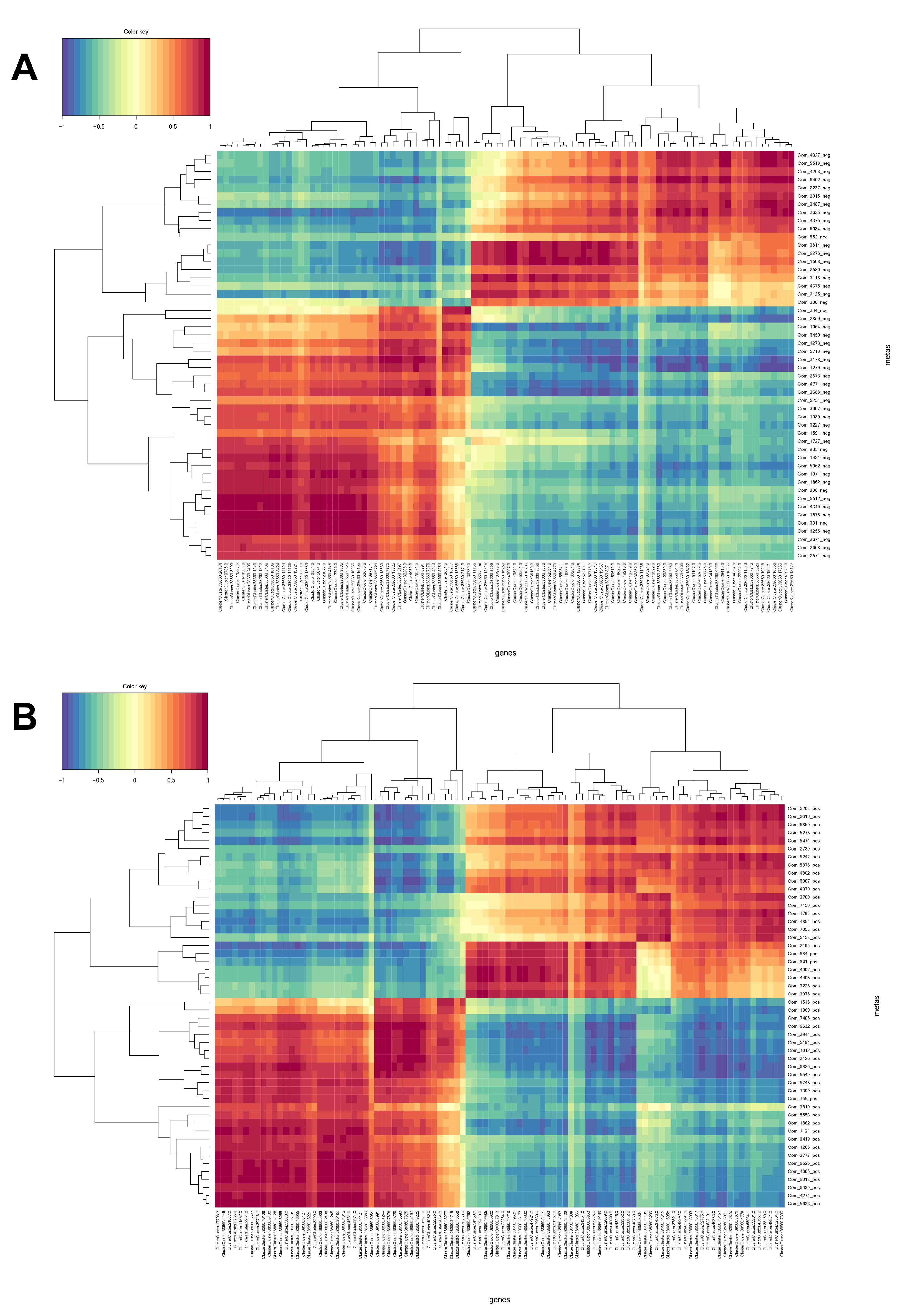
Figure S2 shows that the metabolic pathways of coumarins and flavonoids are not downregulated. Therefore, there is no contradiction between the downregulation of the phenylpropane synthesis pathway shown by transcriptome analysis and the upregulation of coumarin and flavonoid metabolites shown by metabolome analysis. In addition, the integrated analysis of the metabolome and transcriptome showed that there was a significant positive or negative correlation between differential metabolites and differentially expressed genes (
Figure 7). Therefore, the downregulation of genes related to the phenylpropane metabolic pathway is reflected in the downregulation of genes shown in
Figure S2 and may also show that some negatively regulated transcription factor genes are downregulated.
The biosynthesis of secondary metabolites such as phenylpropanes is mainly regulated by transcription factors at the transcriptional level [
29]. Recently, it has been found that six TF families are closely related to defense signaling. These six TF families are: AP2/ERF (APETALA2/ethylene responsive factor), bHLH (basic helix-loop-helix), MYB (myeloblastosis related), NAC (no apical meristem (NAM), Arabidopsis transcription activation factor (ATAF1/2), cup-shaped cotyledon (CUC2)), WRKY, and bZIP (basic leucine zipper) [
30,
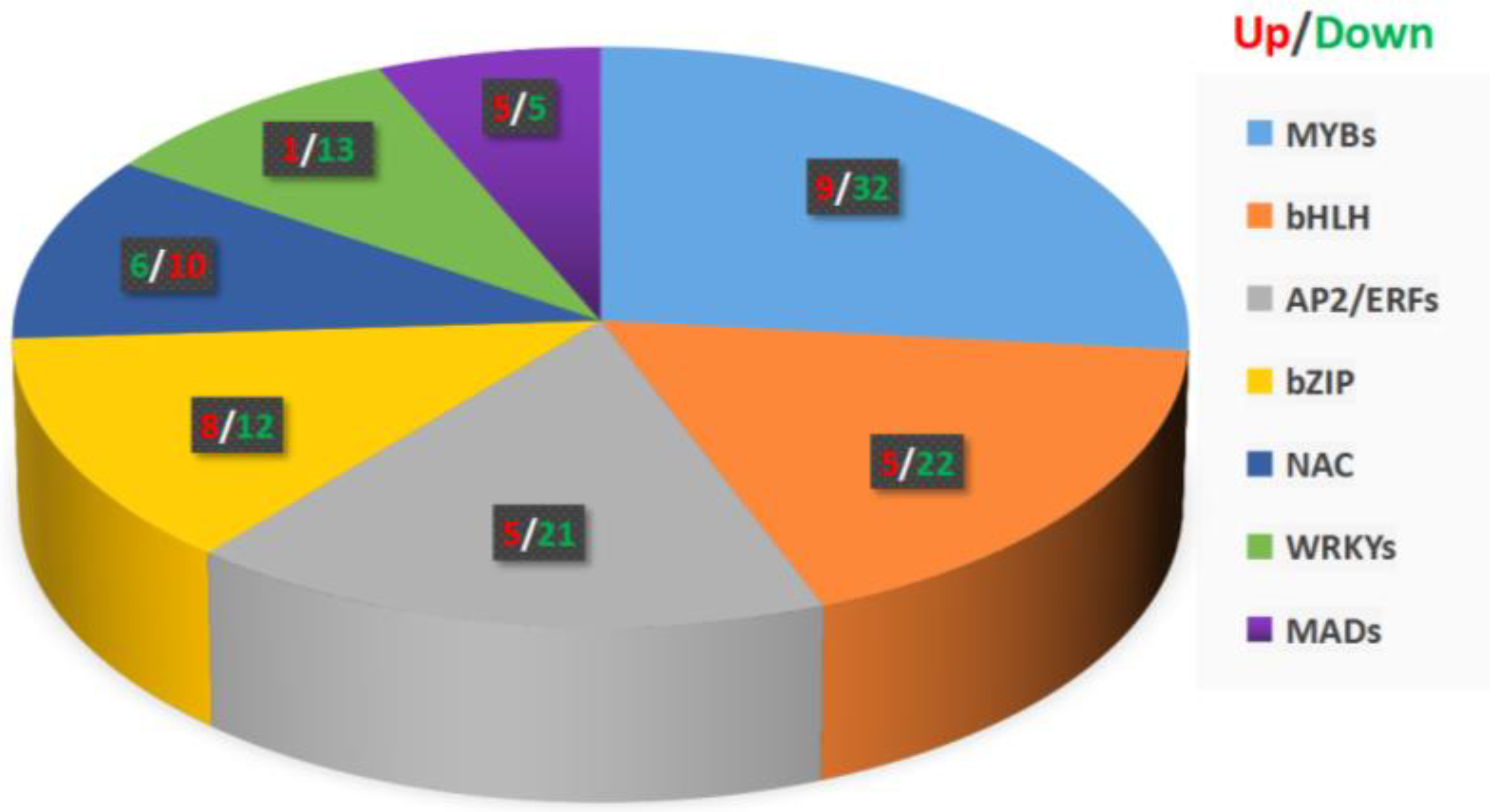
31]. Interestingly, our transcriptome analysis showed that most of the DEGs with |log2FC| ≥ 1 are transcription factor genes. Among them, 41 MYB, 27 bHLH, 26 AP2/ERF, 20 bZIP, 16 NAC, 14 WRKY, 12 AUX/IAA, 10 MAD, and 12 HSF transcription factor genes were found. Clearly, these transcription factors are related to plant hormone signal transduction, stress resistance, and secondary metabolite synthesis. Since most of the transcription factors related to the phenylpropane metabolic pathway shown in
Figure 6 are downregulated, we speculate that the downregulation of these transcription factors leads to the downregulation of the DEGs shown in
Figure S2. It is also possible that some transcription factors play a negative regulation role and the down-regulation of transcription factors promote the coumarin and flavonoid metabolic pathways in the phenylpropane metabolic pathway. In addition, we did not rule out the possibility that the downregulation of other pathways of the phenylpropane metabolic pathway can promote the metabolism of coumarins and flavonoids.
Our previous study found that stress conditions can promote the release of hydrogen in plants [
7]. Hydrogen can regulate the expression of transcription factor genes related to stress and plant hormone signal transduction, which suggests that hydrogen may be the secondary messenger under stress conditions. Since plant stress resistance and the accumulation of secondary metabolites are related to the regulation of plant hormones [
7], these results suggest that hydrogen may act as a signal molecule to regulate the expression of transcription factor genes related to plant hormone and defense signal transduction, which is consistent with our previous research conclusions.
4. Materials and Methods
4.1. Plant Materials and Treatments
Twelve Wuzhimaotao (
Ficus hirta Vahl) plants growing in South China Botanical Garden were selected and evenly divided into two groups, the treatment group and the control group. Hydrogen water was obtained following the method of Li et al. [
32]. Purified hydrogen gas (99.99%,
v/
v) (Kedi Gas Chemical Co., Ltd., Foshan, China) was bubbled into 5 L of pure water at a rate of 200 mL min
−1 for 3 h to obtain the saturated hydrogen water. The hydrogen concentration of hydrogen water was determined using a hydrogen portable meter (Trustlex Co., Ltd., ENH-1000, Yokohama, Japan). The treatment group was watered with hydrogen water (0.8 ppm) once a week for a total of 3 times, while the control group was watered with pure water. On the 15th day, the roots of
Ficus hirta Vahl were collected, frozen with liquid nitrogen and stored at −80 °C before metabolite extraction and RNA-Seq.
4.2. Metabolite Extraction and Analysis
Each sample tissue (100 mg) was grounded with liquid nitrogen, and the homogenate was resuspended with precooled 80% methanol and 0.1% formic acid by well vortexing. The sample was placed on ice for 5 min and then centrifuged at 15,000 rpm at 4 °C for 5 min. Part of the supernatant was diluted with methanol to a final concentration of 60% with LC-MS grade water.
The samples were subsequently transferred to a fresh Eppendorf tube with a 0.22 μm filter and then were centrifuged at 15,000× g at 4 °C for 10 min. Finally, the filtrate was injected into the LC-MS/MS system analysis. Liquid sample (100 μL) and prechilled methanol (400 μL) were mixed by well vortexing.
The Vanquish UHPLC system (Thermo Fisher, MA, USA) and Orbitrap Q Exactive series mass spectrometer (Thermo Fisher, MA, USA) were used for LC-MS/MS analysis. The flow rate of sample injected into a Hyperil Gold column (100 × 2.1 mm, 1.9 μm) was 0.2 mL/min, and the linear gradient retention time was 16 min. Eluent A (0.1% FA in water) and eluent B (Methanol) were used for positive mode. Eluent A (5 mM ammonium acetate, pH 9.0) and eluent B (Methanol) were used for negative polarity mode. The setting of solvent gradient was: 2% B, 1.5 min; 2–100% B, 12.0 min; 100% B, 14.0 min; 100–2% B, 14.1 min; and 2% B, 16 min. Under the positive and negative polarity mode, the Q Exactive mass spectrometer was run with a spray voltage of 3.2 kV, the capillary temperature at 320 °C, a sheath gas velocity of 35arb, and an auxiliary gas velocity of 10arb.
For peak alignment, pickup, and quantification of each metabolite, Compound Discoverer 3.0 (CD3.0, Thermo Fisher) was used to process the raw data file generated by UHPLC-MS/MS. The main parameters were as follows: retention time tolerance was 0.2 min; the actual mass tolerance was 5 ppm; signal strength tolerance was 30%; the signal-to-noise ratio was 3 and minimum intensity was 100,000. Then, the peak intensity was normalized to the total spectral intensity. Normalized data can be used to predict molecular formulas based on molecular ion peaks, additive ions, and fragment ions. Then, in order to obtain accurate qualitative and relative quantitative results, the peaks were matched with the mzCloud (
https://www.mzcloud.org/, accessed on 23 May 2020) and ChemSpider (
http://www.chemspider.com/, accessed on 23 May 2020) databases. Statistical analyses were performed using the statistical software R (R version R-3.4.3), Python (Python 2.7.6 version), and CentOS (CentOS release 6.6). When the data did not conform to the normal distribution, the area normalization method was used for normal transformation.
These metabolites were annotated using the HMDB database (
http://www.hmdb.ca/, accessed on 23 May 2020), KEGG database (
http://www.genome.jp/kegg/, accessed on 23 May 2020), and Lipidmaps database (
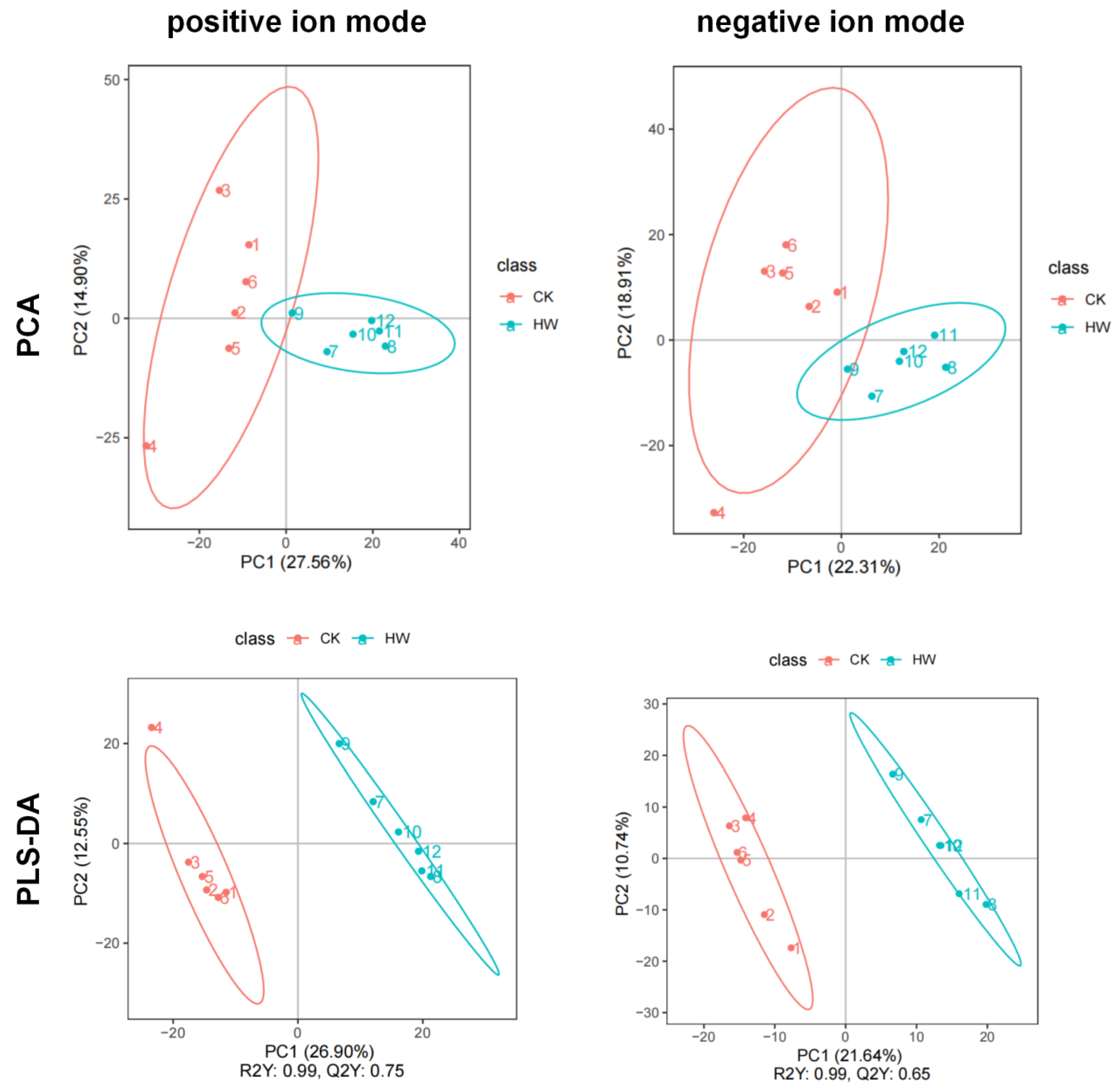
http://www.lipidmaps.org/, accessed on 23 May 2020). Principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) were performed in metaX. Statistical significance (
p-value) was calculated by univariate analysis (
t-test). The identification criteria of differential metabolites were VIP > 1,
p-value < 0.05, and fold change ≥ 2 or FC ≤ 0.5. The metabolites of interest were filtered according to the log2 (FC) and-log10 (
p-value) of metabolites to obtain the volcano map.
The data were normalized using the Z score of the intensity region of differential metabolites, and the clustering heat map was drawn using the phatmap software package in R language. Cor() in R language (method = pearson) was used to analyze the correlation between different metabolites. We used cor.mtest() in R language to calculate the statistically significant correlation between different metabolites. The p-value with statistical significance was <0.05, and the correlation diagram was drawn with the corrplot software package in R language. We used the KEGG database to study the function and metabolic pathways of these metabolites. The criteria for metabolic pathway enrichment of differential metabolites are as follows. The metabolic pathway enrichment of differential metabolites were performed, when ratio were satisfied by x/n > y/N, metabolic pathway were considered as enrichment, when p-value of metabolic pathway < 0.05, metabolic pathway were considered as statistically significant enrichment.
4.3. Total RNA Extraction, RNA Quantification and Qualification
Frozen samples were ground in liquid nitrogen. Total RNA was extracted using a TRIzol reagent. We used 1% agarose gel electrophoresis to monitor RNA degradation and pollution. The NanoPhotometer® spectrophotometer (IMPLEN, Westlake Village, CA, USA) was used to check RNA purity. The RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA) was used to assess RNA integrity.
4.4. Library Preparation for Transcriptome Sequencing
The total amount of RNA in each sample used as input material for RNA sample preparation was 1.5 µg. Following the manufacture’s recommendations and adding index codes to attribute sequences to each sample, a NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA) was used to generate sequencing libraries. In short, mRNA was purified from total RNA by poly-Toligo-attached magnetic beads.
Under elevated temperatures, divalent cations were used for cleavage in the NEBNext First Strand Synthesis Reaction Buffer (5X). The first strand of cDNA was synthesized by random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). The second strand of cDNA was then synthesized using DNA Polymerase I and RNase H. The remaining overhangs were transformed into blunt ends by exonuclease/polymerase activities. After adenylation of 3’ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure were ligated to prepare for hybridization.Library fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, USA) to preferentially select cDNA fragments with a length of 250~300 bp. Then 3 µL of USER Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min followed by 5 min at 95 °C before PCR. PCR was then performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) Primer. Finally, the PCR products were purified (AMPure XP system) on the Agilent Bioanalyzer 2100 system and the library quality was evaluated.
4.5. Clustering and Sequencing (Novogene Experimental Department)
A TruSeq PE Cluster Kit v3-cBot-HS (Illumia) was used to cluster the index-coded samples on a cBot Cluster Generation System according to the manufacturer’s instructions. The prepared libraries were then sequenced on the Illumina Hiseq platform, and paired end reads were generated.
4.6. Data Analysis
Quality control: Raw data (raw reads) in fastq format were processed through the internal Perl script. In this process, clean data (clean reads) were obtained by deleting the reads including adapter, poly-N, and low-quality reads from the raw data. In addition, Q20, Q30, GC content, and sequence repeat level of clean data needed to be calculated. All the downstream analyses were based on clean data with high quality.
Transcriptome assembly: The left files (read1 files) in all libraries and samples were merged into a large left.fq file, and the right files (read2 files) were merged into a large right.fq file. Transcriptome assembly was based on the left.fq and right.fq, and was completed using Trinity [
33]; min_kmer_cov was set to 2 by default, and all other parameters were also set by default.
Gene function annotation: Gene functions were annotated based on Nr (NCBI nonredundant protein sequences), Nt (NCBI nonredundant nucleotide sequences), KOG/COG (Clusters of Orthologous Groups of proteins), Pfam (Protein family), Swiss-Prot, KO (KEGG Ortholog database), and GO (Gene Ontology) databases.
Differential expression analysis: The differential expression of two groups of samples with biological duplication was analyzed by DESeq R package (1.10.1). DESeq provides statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. In order to control the error detection rate, the resulting p value was adjusted using the method of Benjamin and Hochberg. Genes found by DESeq were identified as differentially expressed genes if their adjusted p value was <0.05. The threshold of significant differential expression was set as q value < 0.005 and |log2 (foldchange)| > 1.
GO enrichment analysis: GO enrichment analysis of the differentially expressed genes (DEGs) was realized by the GOseq R packages based on Wallenius’ noncentral hypergeometric distribution [
34].
KEGG pathway enrichment analysis: KEGG [
35] is a database resource (
http://www.genome.jp/kegg/, accessed on 28 December 2021) used to understand the advanced functions and utilities of biological systems, such as cells, organisms, and ecosystems, from molecular level information, especially large-scale molecular data sets generated by genome sequencing and other high-throughput experimental technologies. KOBAS [
36] software was used to test the statistical enrichment of differentially expressed genes in the KEGG pathway.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






