1. Introduction
Cryopreservation is defined as the conservation of germplasm in ultra-low temperatures (−196 °C), normally in liquid nitrogen (LN), thus allowing the preservation of material using a small space while presenting a low contamination risk and requiring minimal maintenance [
1,
2]. In addition, cryopreservation permits the conservation of material for long periods with the assurance of genetic stability since it is believed that all metabolic and physiological processes are arrested under these conditions [
1,
2,
3,
4].
Cryopreservation is emerging as a useful strategy for the long-term and safe conservation of embryogenic cultures (ECs) generated in somatic embryogenesis protocols, as the maintenance of these materials under cryopreserved conditions preserves cell viability without the loss of embryogenic potential [
5]. Indeed, this strategy has been constantly applied for the cryopreservation of somatic embryos of different plant species [
6,
7,
8,
9]. For bamboo, an important group of the grass family, somatic embryogenesis represents a suitable strategy of in vitro propagation as it solves difficulties faced by conventional propagation methods, such as damage to the mother plant and irregular or not always viable seed production [
10]. The association of this technique with cryopreservation represents a perfect match for bamboo propagation and conservation; however, no reports exploiting cryopreservation and somatic embryogenesis have been described [
11]. Only two reports on the slow-growth conservation of somatic embryos of Dendrocalamus hamiltonii [
12,
13] and one on cryopreservation of the whole zygotic seed [
14] were available to date, demonstrating the need for new strategies for the in vitro conservation of bamboo species.
Guadua chacoensis, the species studied in this work, is a lignified bamboo (
Bambusoideae) presenting natural distribution in Argentina, Bolivia, Brazil, and Paraguay [
15]. Its seed production occurs irregularly and in long cycles of approximately 28 years [
16]. The use of in vitro biotechnology tools such as somatic embryogenesis and cryopreservation are fundamental for its propagation and conservation.
Over more than 60 years of using cryopreservation, various methodologies have been established, i.e., classical slow freezing, encapsulation/ dehydration, vitrification, droplet vitrification, and D and V cryo-plate methods [
2]. The classical slow-freezing method, also known as the two-step freezing technique, includes pre-growth with osmotic agents to acclimate cells and increase freezing tolerance, and the cryoprotective treatment. Additionally, during the freezing step, the use of a Mr. Frosty unit allows for the slow cooling of the material, providing a decrease of 1 °C per min until a temperature of −40 °C is achieved. This is followed by direct immersion in liquid nitrogen [
5,
17,
18]. Overall, the primary purpose of cryopreservation, independent of the methodology applied, is to control dehydration in order to avoid cryoinjuries during the freezing and thawing steps [
18].
Low temperatures during the cryopreservation process can impose stress on plant cells, including physical damage, changes in membrane viscosity, retardation of metabolic activity, and the formation of radicals that lead to oxidative stress [
19]. Among the physical damages, the most critical effect is the disruption of membranes and disconnection of the cells as a consequence of intracellular ice crystal formation [
2]. One way to prevent this is by using cryoprotective agents (CPAs) that can permeate cell membranes, including DMSO and glycerol, and non-permeable agents, including polyethylene glycol and sucrose [
5]. The exact method by which CPAs act in the cell is still unclear. However, it is suggested that CPAs, particularly the permeable ones, are able to increase cell osmolarity and reduce cell water content, resulting in better cell tolerance to dehydration [
17]. Moreover, the use of these agents may facilitate the transition of the water phase directly to the vitrification phase, also known as the glassy state, without the formation of ice crystals [
2].
The generation of reactive oxygen species (ROSs), such as hydrogen peroxide (H
2O
2), superoxide radical (O
2•−), hydroxyl radical (OH
•), and singlet oxygen (
1O
2) [
20], is inevitable during the multiple steps of cryopreservation [
21,
22]. An excessive imbalance between ROS production and the activity of antioxidant enzymes, such as superoxide dismutase (SOD), catalase (CAT), ascorbate peroxidase (APX), and guaiacol peroxidase (GPX), could result in oxidative stress, which disrupts cellular homeostasis and can lead to membrane damage by lipid peroxidation, protein oxidation, DNA damage, and, in the most serious case, results in programmed cell death [
22]. In addition, high oxidative stress can negatively affect cryopreservation and might lead to reduced culture viability and recovery rates [
9,
23,
24,
25].
In our previous study, we described and characterized the somatic embryogenesis of
Guadua chacoensis using morphohistological and biochemical features [
26]. Therefore, the present study aims to establish a cryopreservation protocol for
G. chacoensis ECs using the classical two-step freezing procedure and to characterize the role of antioxidant enzymes in the cryopreservation process.
3. Material and Methods
3.1. Plant Material
Embryogenic cultures (ECs) of
G. chacoensis, obtained as previously described by Giacomolli Polesi et al. [
26], were used as plant material for the establishment of suspension cultures. To establish the suspension cultures, 300 mg of ECs were inoculated in 250 mL Erlenmeyer flasks containing 50 mL of suspension culture medium (SCM), which was composed of MS basal salts [
27] supplemented with L-glutamine (1 g L
−1), sucrose (30 g L
−1), Morel vitamins (2 mL L
−1) [
28], and Picloram (10 μM). The pH of the culture medium was adjusted to 5.8 before autoclaving at 121 °C and 1.5 atm for 15 min. The Erlenmeyer flasks were kept in dark conditions, using an orbital shaker to achieve permanent agitation (95 rpm), in the growth room at 25 ± 2 °C for 45 days.
3.2. Analysis of Cell Growth Dynamics of Suspension Cultures
To characterize the growth phases of
G. chacoensis ECs (lag, exponential, or logarithmic, linear, and decline), two different parameters were analyzed: a destructive method, measuring the ECs’ fresh weight; and a non-destructive method, measuring the cell volume after sedimentation using a device developed for suspension-culture sedimentation with a ruler, proposed by Mustafa et al. [
18]. Thus, 24 replicates of a 250 mL Erlenmeyer flask containing 300 mg of
G. chacoensis EC and 50 mL of SCM were used. Both measures were performed every 7 days until day 56 of culture, when the growth ratio was obtained using the following equations: (i) fresh weight at day x/the initial fresh weight; and (ii) cell volume after sedimentation at day x/initial cell volume after sedimentation.
3.3. Phytotoxicity Evaluation of Cryoprotective Solution
Following the methodology proposed by Mustafa et al., a phytotoxicity test was utilized to evaluate the possible toxicity of a cryoprotective solution for
G. chacoensis ECs [
18]. The cryoprotective solution was composed of 2 M of sucrose, 1 M of glycerol, 1 M of dimethylsulfoxide (DMSO), and 1% proline (weight: volume). The final two components were filter esterized in a flow chamber in the cool autoclaved solution (121 °C, 1.5 atm for 12 min). In this bioassay, ECs were subjected to five incubation times (0, 30, 20, 120, and 240 min) without being subjecting to the freezing step, as was proposed by Mustafa et al. [
18] and Fraga et al. [
7], with modifications. Four replications of the Erlenmeyer flasks, each containing 1 g of
G. chacoensis ECs and 50 mL of SCM at day 39 of subculture, were used for each incubation time, totaling 20 Erlenmeyer flasks.
Briefly, 25 mL of SCM was removed and replaced by the same volume of pretreatment solution, which consisted of the basal SCM medium (Picloram-free) plus mannitol (180 g L−1). The suspension cultures were pre-incubated for two days in an orbital shaker at 95 rpm and 25 ± 2 °C in dark conditions. The cultures were then kept in the device until the sedimentation of the cells and the removal of the culture medium above the cells (approximately 25 mL). A cold, cryoprotective solution (0 °C) was added in the same volume that was removed, and the five incubation times were tested.
The incubation process was performed in an orbital shaker at 95 rpm and 0 °C. We then used a sterilized funnel, attached to a Kitasato flask with a vacuum pump, for the suction of the liquid medium and collection of the G. chacoensis ECs. These cultures were transferred to Petri dishes containing two filter-paper disks and 25 mL of MSC medium gelled with agarose (7.5 g L−1). After 2 days of incubation, the upper filter paper with the EC was subcultured to a fresh SCM medium gelled with agarose (7.5 g L−1), and. After 7 days, the cultures were transferred to SCM gelled with phytagel (2 g L−1) without filter paper. The cultures were kept in constant darkness at 22 ± 2° C.
The G. chacoensis ECs were weighted at 4, 11, 20, and 30 days of culture, and the final weight-increased ratio after the phytotoxicity test was obtained using the following equation: fresh weight at day 30/fresh weight at day 4.
3.4. Cryopreservation Experiments
After evaluating the toxicity of the cryoprotective solution and defining the best incubation times, the complete cryopreservation protocol was performed according to Mustafa et al. [
18] and Fraga et al. [
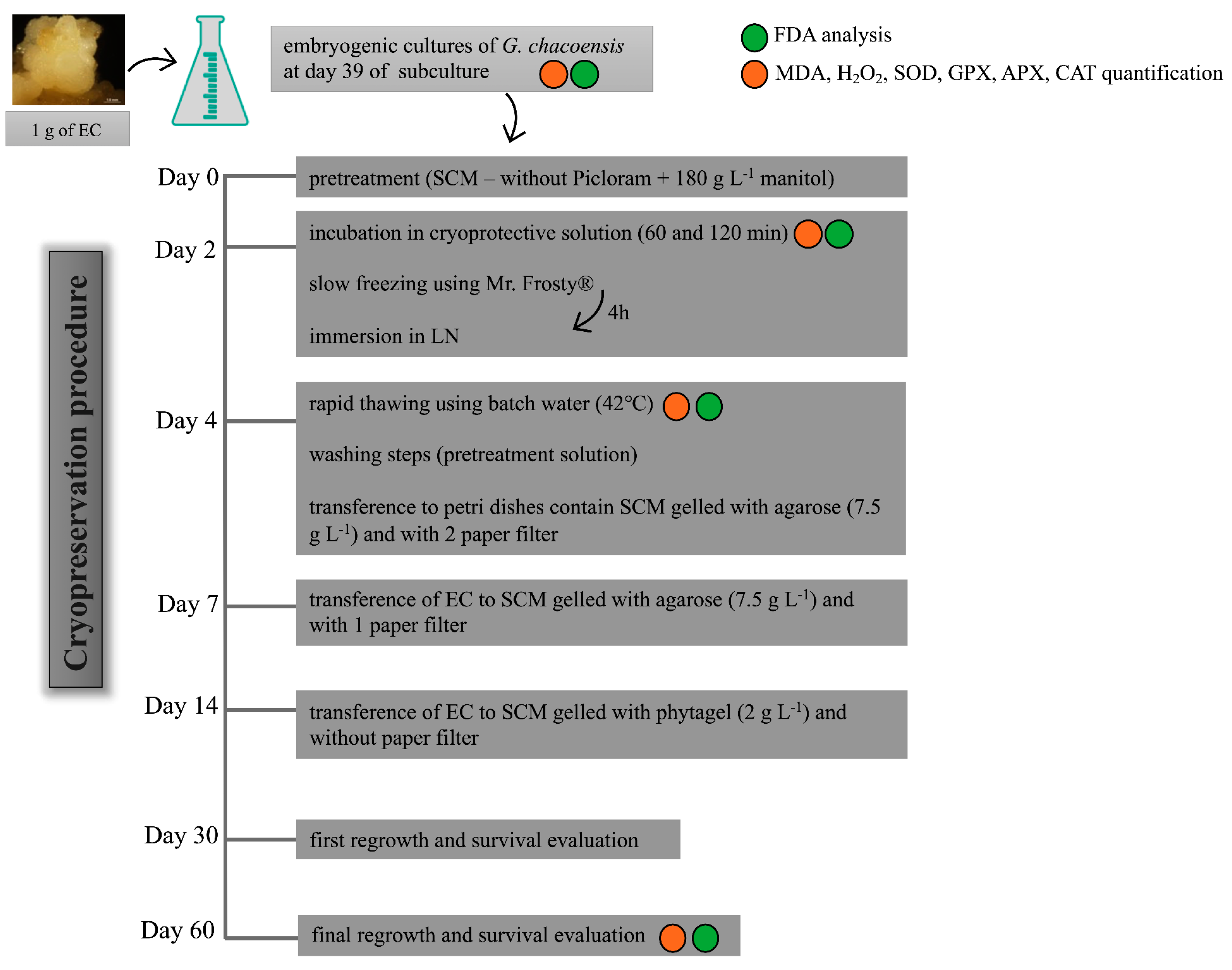
7], with adaptations. The step-by-step cryopreservation procedure used in the present study is shown in
Figure 7.
G. chacoensis ECs, at 39 days of culture and in the exponential phase, were used as plant material. The experiment was realized using five replicates for each incubation time, totaling 15 Erlenmeyer flasks. The initial steps of the protocol were the same as described above, whereas the suspension cultures were first incubated for 2 days in a pretreatment solution in an orbital shaker at 95 rpm and 25 ± 2 °C in dark conditions. This was followed by incubation in the cryoprotective solution for 60 and 120 min in an orbital shaker at 95 rpm and 0 °C. After this period, the G. chacoensis ECs were collected in a sterilized funnel with a filter paper attached to a Kitasato flask, using a vacuum pump for the suction of the liquid medium and cryoprotective solution. Next, approximately 300 mg of the collected ECs was transferred to each cryovial (2 mL), totaling 18 cryovials from each incubation time. These were transferred to Mr. Frosty (Nalgene®, Waltham, MA, USA) containers containing 250 mL of isopropanol. The containers were maintained in an ultra-freezer (−80 °C) for 4 h for controlled cooling according to the manufacturer’s instructions. Afterward, the cryovials were immediately transferred to liquid nitrogen and were kept immersed for at least 24 h.
The subsequent step involved the rapid thawing of the cultures through the immersion of the cryovials in a deionized water bath at 40 °C for approximately 2 min. The cryotubes were kept on ice for a few minutes until the cultures decanted to the bottom of the tubes. Thus, the superior phase without cultures was removed and replaced by the same volume (1 mL) of washing solution (pretreatment solution—SCM plus mannitol (180 g L−1)). Next, the cultures were decanted again, the washing solution was discarded, and the cultures were transferred to Petri dishes containing two filter-paper disks and 25 mL of SCM medium gelled with agarose (7.5 g L−1). After 3 days of incubation, the upper filter paper with the G. chacoensis cultures was subcultured to a fresh SCM medium gelled with agarose. Subsequently, after 7 days, the cultures were transferred to SCM gelled with phytagel (2 g L−1) without filter paper. The cultures were kept in constant darkness at 22 ± 2 °C for 60 days.
The EC regrowth was evaluated at 30 and 60 days after the cryopreservation procedure for each incubation time. A control treatment, which consisted of a cell suspension of G. chacoensis ECs that were not subjected to the cryopreservation protocol, was used as a control for the regrowth analysis. The experiment was carried out in a completely randomized design, with six Petri dishes containing three colonies of G. chacoensis ECs each, and the regrowth rate was evaluated using the following equation: fresh weight at day 30 or 60/initial fresh weight.
For the quantification of the MDA and H
2O
2 content and the determination of antioxidant enzyme activity (the activity of SOD, CAT, APX, and GPX), samples were collected in three different stages of the cryopreservation protocol: (i) after incubation in cryoprotection solution; (ii) after freezing and thawing; and (iii) 60 days after regrowth (
Figure 7). Controls corresponding to the suspension culture that were not subjected to the cryopreservation procedure for steps (i) and (ii) and this same material were collected, transferred to Petri dishes on day 4, and then passed through all the following steps until day 60 regrowth (
Figure 7), corresponding to the control of step (iii).
3.5. Quantification of MDA and H2O2 Content
The quantification of the MDA and H
2O
2 content was performed according to Velikova et al. [
29], with an adaptation described by Polesi et al. [
30].
Briefly, three samples of 200 mg of fresh material were collected from each incubation time (60 or 120 min) and the control treatment. The extraction procedure was performed through the maceration of the samples, followed by homogenization with 0.1% trichloroacetic acid (TCA) (1/5; w/v) and centrifugation at 12,000 rpm and 4 °C for 20 min. The supernatant collected was used as a culture substrate in the reaction mixture for the determination of the MDA and H2O2 content.
Contents of MDA were quantified using the thiobarbituric acid (TBA) method, in which 200 μL of the culture extract was mixed with 400 μL of a buffer containing 20% (w/v) trichloroacetic acid (TCA) and 0.5% (w/v) TBA and incubated in bath water at 95 °C for 30 min. After that, the microtubes were placed on ice for 15 min, followed by centrifugation at 10,000 rpm for 15 min at 4 °C and the subsequent collection of the supernatants. The MDA concentration was determined by reading 200 μL of the mixture in a spectrophotometer (SpectraMax Paradigm, Molecular Devices, San Jose, CA, USA) at 532 nm, adjusted for nonspecific absorbance at 600 nm using an extinction coefficient of 155 mM cm−1. The contents were expressed in μmol g−1 of fresh material.
For the quantification of H2O2 contents, 200 μL of the culture extract was mixed with 100 μL of 10 mM potassium phosphate buffer (pH 7.0) and 200 μL of 1 M potassium iodide and kept for 15 min at room temperature for the activation of the reaction. The H2O2 content was then determined by reading 200 μL of the mixture in a spectrophotometer (SpectraMax Paradigm, Molecular Devices, San Jose, CA, USA) at 350 nm, and the concentration was assessed using a known, standard curve of H2O2. The contents were expressed in μmol g−1 of fresh material.
Using GraphPad Version 8.1.1, data obtained for the MDA and H2O2 content were submitted to a one-way analysis of variance (ANOVA) and the Tukey mean separation test (p < 0.05).
3.6. Antioxidant Enzymes Activity
The determination of the activity of antioxidant enzymes superoxide dismutase (SOD), catalase (CAT), ascorbate peroxidase (APX), and guaiacol peroxidase (GPX) was realized following the methodology proposed by Rao et al. [
31], with adaptations. For this determination, three samples of 200 mg of fresh material were collected from each incubation time (60 and 120 min) and the control treatment. For the extraction procedure, samples were macerated and then homogenized with an extraction buffer composed of 50 mM potassium phosphate buffer (pH 7.0) (1/5;
w/
v), followed by centrifugation at 12,000 rpm for 20 min at 4 °C. The supernatant was collected and used as a culture substrate in the reaction mixture for the determination of the activity of each enzyme.
The SOD (EC 1.15.1.1) activity was determined using 80 μL of culture substrate mixed with 2 mL of 50 mM potassium phosphate buffer (pH 7.8) containing 10 mM methionine, 56 μM nitroblue tetrazolium, and 24 μL of 100 mM riboflavin. The reaction mixture was made in duplicate samples, in which one sample was maintained under dark conditions to be used as a blank and the other was illuminated for 15 min in a box line with aluminum foil (using a 15 W white lamp at a distance of approximately 12 cm from samples). The SOD activity was determined by the reading at 560 nm using a standard curve, which was constructed using purified SOD as the standard.
The CAT (EC 1.11.1.6) activity was determined using 20 μL of culture extract mixed with 180 μL of 50 mM potassium phosphate buffer (pH 7.0) containing 13.3 mM hydrogen peroxide. The CAT activity was determined at 240 nm by the absorbance decreases for 3 min at 25 °C, using the molar extinction coefficient of hydrogen peroxide (39.4 mM cm−1).
The APX (EC 1.11.1.11) activity was determined using 40 μL of culture extract mixed with 160 μL of potassium phosphate buffer (pH 7.0) containing 1.5 mM ascorbic acid and 2 mM hydrogen peroxide. The APX activity was determined at 290 nm by the absorbance decreases for 2 min at 25 °C, using the molar extinction coefficient of ascorbic acid (2.8 mM cm−1).
The GPX (EC 1.11.1.7) activity was determined using 7 μL of culture extract (diluted in extraction buffer 1:80, v/v) mixed with 193 μL of 10 mM sodium phosphate buffer (pH 6.0) containing 0.125% (v/v) H2O2 and 0.250% (v/v) guaiacol. The GPX activity was determined at 470 nm by the absorbance decreases for 3 min at 30 °C, using the molar extinction coefficient of guaiacol (25.2 mM cm−1).
Protein contents were quantified by the Bradford method [
32] at 595 nm, using bovine serum albumin (BSA) as a standard. The activities of SOD, CAT, APX, and GPX were expressed in μKatal. mg
−1 of protein (1 katal signifies the amount of enzyme that catalyzes 1 μmol of a substrate in 1 min).
3.7. Fluorescein Diacetate (FDA) Analysis
FDA staining was used to monitor cell viability during and following the cryopreservation procedure. A stock solution of 1% (weight: volume–FDA: acetone) was prepared and to 2% (volume: volume) in water. Approximately 10 μL of the diluted solution was added directly to approximately 30 mg of EC per 5 min. This was followed by the observation of green fluorescence under UV illumination using an inverted microscope (Olympus IX81) coupled with a digital camera (Olympus DP71, Shinjuku, Tokyo, Japan) and the software CellSens Dimension (Olympus). Three representatives and independent samples were analyzed in the following cryopreservation steps: (i) after incubation in cryoprotection solution; (ii) after freezing and thawing; (iii) 60 days after regrowth.
3.8. Statistical Analysis
All the statistical analyses were performed using GraphPad Prisma Version 8.1.1. Data obtained for the increased ratio in phytotoxicity test, regrowth rates, and the quantification of MDA, H2O2, SOD, CAT, APX, and GPX were subjected to a one-way analysis of variance (ANOVA), followed by Tukey mean separation test (p < 0.05). Linear regression was used to evaluate the results of the phytotoxicity test. A correlation analysis was used to estimate the interactions between the data obtained for the quantification of MDA, H2O2, SOD, CAT, APX, and GPX.
4. Discussion
4.1. Cryopreservation Procedure
For several plant species, embryogenic cultures can be efficiently conserved for a long time without the loss of embryogenic potential through the cryopreservation technique. However, there are no reports on the cryopreservation of the somatic embryos of bamboo species, which may be a consequence of the complexity of somatic embryogenesis, the few somatic embryogenesis protocols established, and the scarce information available about this group of plants [
11]. Based on this, the need for studies to establish strategies for the in vitro conservation of bamboo is evident, especially based on cryopreservation approaches.
Cryopreservation is a complex process wherein the achievement of high survival rates depends on the optimization of many factors, including the correct choice of explant. The standardization of the culture’s conditions and the use of appropriate intervals between subcultures can be determinant factors for the success of cryopreservation [
33]. It is suggested that cultures in the exponential phase, in active growth, and which exhibit cells with a low water content, least volume, small vacuoles, and dense cytoplasm, resulting in higher cytosol: vacuole ratio and thus a greater solute concentration, present an improved tolerance to the freezing process [
5,
18,
34]. Our growth dynamics results revealed that the embryogenic cultures of
G. chacoensis are in the exponential/log phase for days 35 to 45 of culture, suggesting that this is the best period for the cryopreservation protocol. However, this behavior should be observed case by case, since each species presents a different growth curve, reinforcing the importance of the analysis of cultures’ growth dynamics before cryopreservation.
Different methodologies can be applied for EC cryopreservation, including two-step, slow-freezing, vitrification, and encapsulation/dehydration [
2]. The choice of the technique depends on the material type and characteristics. Among these methodologies, the most recommended for the cryopreservation of an embryogenic callus suspension culture is the two-step, slow-freezing method, which has been widely reported for species of conifers [
7,
8,
35], bananas [
36], and herbaceous and hardwood species [
5]. No works have been reported for bamboos species, emphasizing the importance of researching cryopreservation strategies for this group of plants. The purpose of the use of the two-step, slow freezing protocol is to: a) expose the cells to a high-concentration solution to stimulate dehydration and b) to use appropriate cooling conditions and cryoprotective agents in such a manner that the intracellular content becomes viscous and forms the so-called “glassy state”, avoiding the formation of harmful crystal ice [
37].
Therefore, the present study established a two-step, slow-freezing cryopreservation protocol for the suspension of cultures of G. chacoensis ECs using preincubation in a culture medium with an osmotic agent (mannitol 180 g L−1) for two days, followed by incubation in a cryoprotective solution with four kinds of cryoprotective agents: sucrose (2 M), glycerol (1 M), DMSO (1 M; 7.1%), and proline (1%), and slow-cooling using Mr. Frosty per 4 h before immediate immersion in LN.
4.2. Cryoprotective Agents and Their Importance for Increasing Cell Tolerance to Dehydration and Freezing Steps
The choice of cryoprotective, the concentration, and the time of incubation used are germplasm-dependent and should be optimized to avoid possible toxic effects that can affect cell viability. Cryoprotective agents (CPAs) facilitate cell cytosol concentration, avoid ice crystal formation, and increase cell tolerance to dehydration and freezing [
17]. Indeed, the purpose of using these CPAs is to properly combine them with the optimal cryopreservation methodology so that ice formation is negligible [
17]. Two types of cryoprotective agents can be used in cryopreservation procedures: ones which are permeable (penetrating) for the membrane, including DMSO, glycerol, and non-ionic molecules, and ones which are not permeable (non-penetrating) for the membrane, including polyethylene glycol and sucrose [
37]. It is suggested that the combination of more than one agent in a lower concentration, as was used in our work, appears to be more appropriate than the use of only one agent in a high concentration for achieving higher viability and cell survival [
5]. Thus, optimizing the CPA concentration and time of exposure is crucial, as toxicity and permeability seem to be germplasm-dependent [
17].
Our phytotoxicity test results revealed that the incubation of
G. chacoensis ECs for 30 and 60 min in a cryoprotective solution did not affect their growth dynamics. However, incubation for 120 min was deleterious after the twentieth day in culture, and incubation for 240 min was phytotoxic. Inversely, for the
Araucaria angustifolia embryogenic cell lines, no inhibitory effects were observed with the same cryoprotective solution used in our study, even with an incubation period of 240 min [
7]. Indeed, the time of exposure varies among species, and optimization is necessary for the success of the cryopreservation procedure. Salaj et al. [
35] reported a 60 min incubation for embryogenic cultures of
Pinus Nigra using a cryoprotective solution with 18% sucrose and 7.5% DMSO, followed by slow-freezing, while Lambardi et al. [
5] proposed that 30 min of incubation presented the highest callus regrowth when compared to 60 and 90 min of incubation in the two-step freezing cryopreservation of Cypress embryogenic masses using a solution containing 180 g·L
−1 of sucrose and 7.5% DMSO; they also reported that conversion rates were not affected when compared to the control. Based on our phytotoxicity test, we used 60 and 120 min incubation times in our further cryopreservation procedure.
4.3. Regrowth Rates and Cell Viability after Cryopreservation
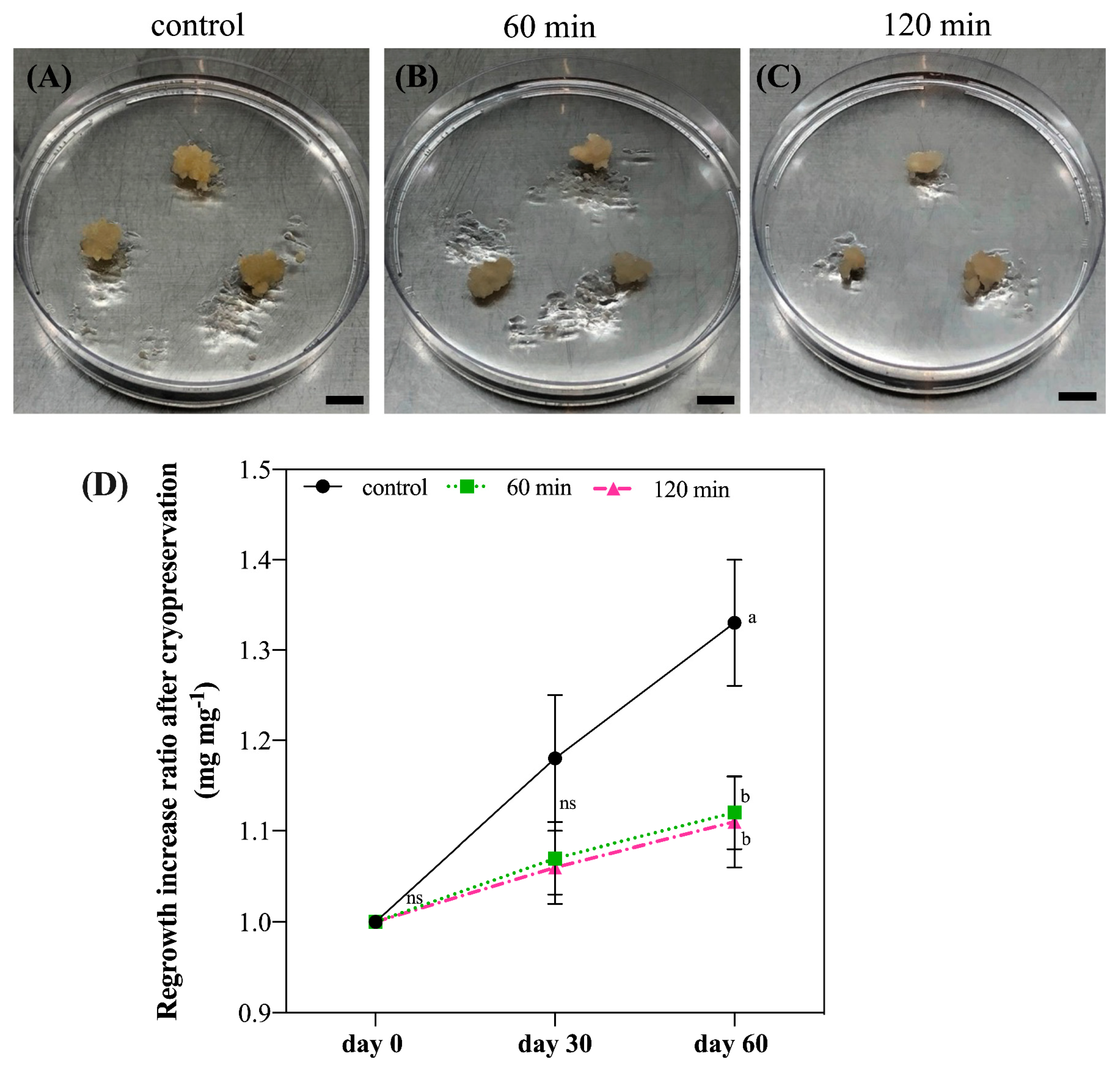
The analysis of regrowth after cryopreservation revealed a 100% cell survival rate, independent of the time of incubation (60 or 120 min), and the cultures presented a 10% increase ratio in fresh material at 60 days after cryopreservation. In fact, increased fresh material may be a good indicator of cell re-establishment after cryopreservation [
34]. In addition, the results of our FDA analysis showed that, although
G. chacoensis ECs may suffer some stress during the incubation in the cryoprotective solution, the viability of the cells was maintained. Additionally, the highest viability was observed 60 days after regrowth, suggesting that our protocol allowed for the successful cryopreservation of ECs of
G. chacoensis. The FDA test is commonly used to verify cell viability during cryopreservation [
7,
18,
34,
35]. In this assay, cell wall esterase cleaves fluorescein diacetate and fluoresce green. In this way, only cells with intact plasma membranes are able to fluoresce, while non-viable cells appear colorless [
38].
Previous studies have shown that the regrowth and conversion rates after cryopreservation were directly affected by the time of exposure to cryoprotectant treatment and the kind of cryoprotectant used. Delgado-Aceves et al. [
39] revealed that the regrowth and conversion of
Agave somatic embryos achieved higher rates after exposure for 15 and 30 min in a plant vitrification solution (composed of glycerol, DMSO, ethylene glycol, and sucrose) when compared to exposures of 45 and 60 min, suggesting that lower incubation times in pretreatment solution may result in major recovery and conversion rates. Salaj et al. [
40] indicated that the regrowth rate after cryopreservation of ECs of different cell lines of
P. nigra were affected by the pre-treatment used, in which sorbitol and maltose pre-treatment resulted in less regrowth than sucrose and cryopreservation did not affect the conversion and proliferation of cultures. In our study, we established a successful cryopreservation protocol with 100% survival rates using both 60 and 120 min incubation. However, there is still a gap in the maturation and conversion phases of
G. chacoensis ECs, showing that further studies should be applied to optimize these steps.
One of the possible reasons for the success of our cryopreservation procedure is the correct establishment of important critical factors, such as the choice of the explant growth period, the use of an efficient pre-treatment, a suitable combination of cryoprotective agents, a suitable time of exposure, an efficient thawing procedure, and the selection of the cryopreservation methodology (slow-cooling using Mr. Frosty). Moreover, our study represents important progress in the cryopreservation of bamboo, being the first work to describe a complete protocol for the cryopreservation of embryogenic cultures of a bamboo species.
4.4. CAT and APX as Central Antioxidant Enzymes in Maintaining G. chacoensis Embryogenic Cultures’ Cell Redox Homeostasis, Assuring Successful Cryopreservation
Cryopreservation is a process that imposes significant stress on cells. Monitoring the activity of antioxidant enzymes during cryopreservation may provide a better comprehension of plant metabolism and response to the production of ROSs [
21,
22]. The analysis of the redox state realized in the present study revealed that the incubation of
G. chacoensis ECs in the cryoprotectant solution resulted in a considerable increase in MDA content, independent of the time of incubation used—60 or 120 min—in addition to a high H
2O
2 content and CAT activity. Interestingly our correlation analysis showed a strong positive correlation between MDA and H
2O
2 and MDA and CAT, proposing a relationship between the data.
Our result of an increased MDA content following incubation in the cryoprotective solution may suggest the occurrence of oxidative stress, implying that this is a critical step in the cryopreservation of
G. chacoensis ECs. As a result of oxidative stress, plant cells may have two different responses: (1) the activation of the antioxidant enzymatic defense system and (2) an increase in lipid peroxidation; for cells survival after cryopreservation, the first reaction is positive and the second is negative [
9]. In fact, previous studies have associated an increase in MDA content—the product of lipid peroxidation—with oxidative stress and the generation of ROSs during different steps of cryopreservation [
9,
41,
42,
43]. Additionally, excessive MDA production is recognized as one of the main factors responsible for low viability after cryopreservation [
9,
23,
24,
25].
Enhanced levels of H
2O
2 found in our work may be associated with oxidative stress as well as with the low activity of SOD, since SOD is the primary enzyme in the H
2O
2 scavenging [
44]. Low SOD activity and high MDA content can negatively affect the survival of cultures after cryopreservation, leading to a higher occurrence of cell ultrastructure damages, i.e., plasmolysis, rupture of the nuclear envelope, and condensation of mitochondria [
42]. Moreover, Ren et al. [
45] suggested that H
2O
2 was the principal ROS molecule involved with oxidative stress during the cryopreservation of
Arabidopsis seedlings, but an enhancement in CAT activity could efficiently scavenge them. Nevertheless, it seems that the ECs of
G. chacoensis may respond to oxidative stress by the activation of CAT, the antioxidant enzyme which presents the highest turnover rate for H
2O
2 dismutation (6 × 10
6 molecules per min) [
44].
In the following step evaluated, after thawing, our results demonstrated a decrease of 40% and 50% in the MDA and H2O2 content, respectively, together with an increase in CAT and APX activities. In addition to this, correlation analysis revealed a positive correlation between MDA and H2O2 and CAT and APX and an inverse negative correlation between H2O2 and CAT, proposing that CAT and APX may be acting as efficient ROS scavengers, resulting in the reduction of oxidative stress and, consequently, decreases in the content of H2O2 and MDA, even after freezing and rewarming steps, which were normally critical steps in cryopreservation procedures.
The importance of the activation of antioxidant enzymes for ROS scavenging during cryopreservation is well known. Viana et al. [
41] have shown that an enhancement in CAT and APX after osmoprotective treatment in the cryopreservation of shoot tips of
Passiflora suberosa was decisive for H
2O
2 scavenging. Chen et al. [
46] proposed that an increase in CAT activity seems to be related to cryogenic stress tolerance and results in increased recovery rates for cryopreserved
Arabidopsis seedlings. James Antony et al. [
47] suggested that the low regeneration rates obtained for cryopreservation by vitrification of the orchid
Dendrobium seem to be associated with the low activity of CAT, POX, and APX during critical steps of the protocol. Poobathy et al. [
48] also proposed that low CAT activity after the post-thawing stages resulted in low survival rates of cryopreserved, protocorm-like bodies of
Dendrobium. An increased activity of APX and CAT during the dehydration and cryostorage treatment steps in the cryopreservation of the orchid
Brassidium indicated the occurrence of high oxidative stress levels [
49]. Indeed, these studies support the hypothesis that the activation of the antioxidant enzymes CAT and APX may be crucial for safeguarding cellular homeostasis in response to the oxidative stress generated after the incubation of G.
chacoensis ECs in the cryoprotective solution. In the same way, the increased activity of antioxidant enzymes also assures enhanced cold tolerance for the cultures, as these enzymes function as efficient ROS scavengers that protect the cell from the excessive ROSs produced in low-temperature conditions, as mentioned by Baek and Skinner [
50].
In the regrowth phase, no significant difference was observed for either antioxidant enzyme activity or the MDA and H2O2 content between the cryopreserved materials and the control, suggesting that although oxidative stress was observed in the initial steps of cryopreservation protocol, a complete recovery of the cryopreserved ECs of G. chacoensis was achieved 60 days after regrowth. Therefore, the activation of the antioxidant enzymes CAT and APX seems to be crucial in safeguarding cell redox homeostasis and assures the maintenance of cell viability after cryopreservation.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}