
Glassy Carbon Electrode Modified with C/Au Nanostructured Materials for Simultaneous Determination of Hydroquinone and Catechol in Water Matrices

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Electrochemical Measurements
2.3. Simultaneous Determination of HQ and CT in Aqueous Matrices by Univariate and Multivariate Calibration
3. Results and Discussion
3.1. Determination of HQ Using GCE Modified with Different MWCNTs
3.2. Accumulation Time Effect on the Oxidation of HQ
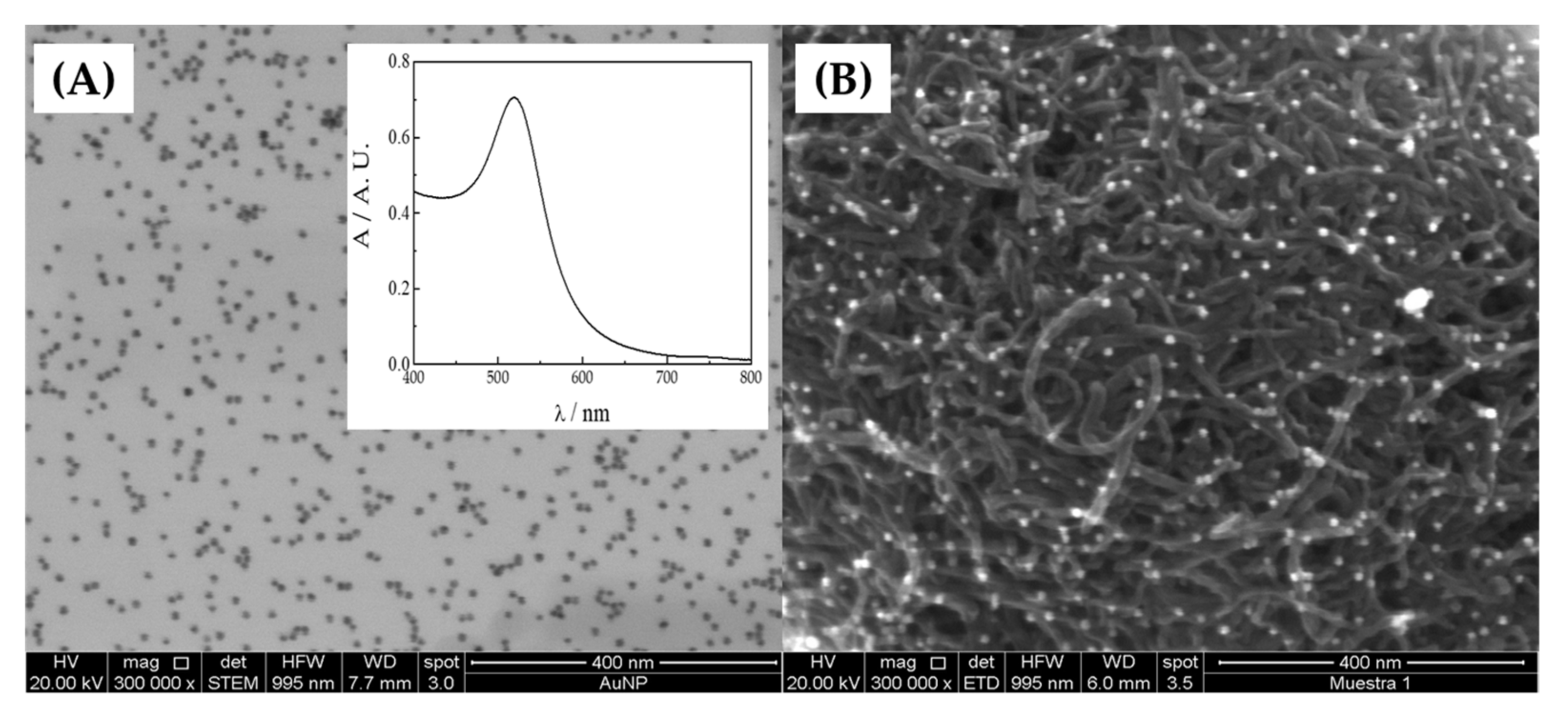
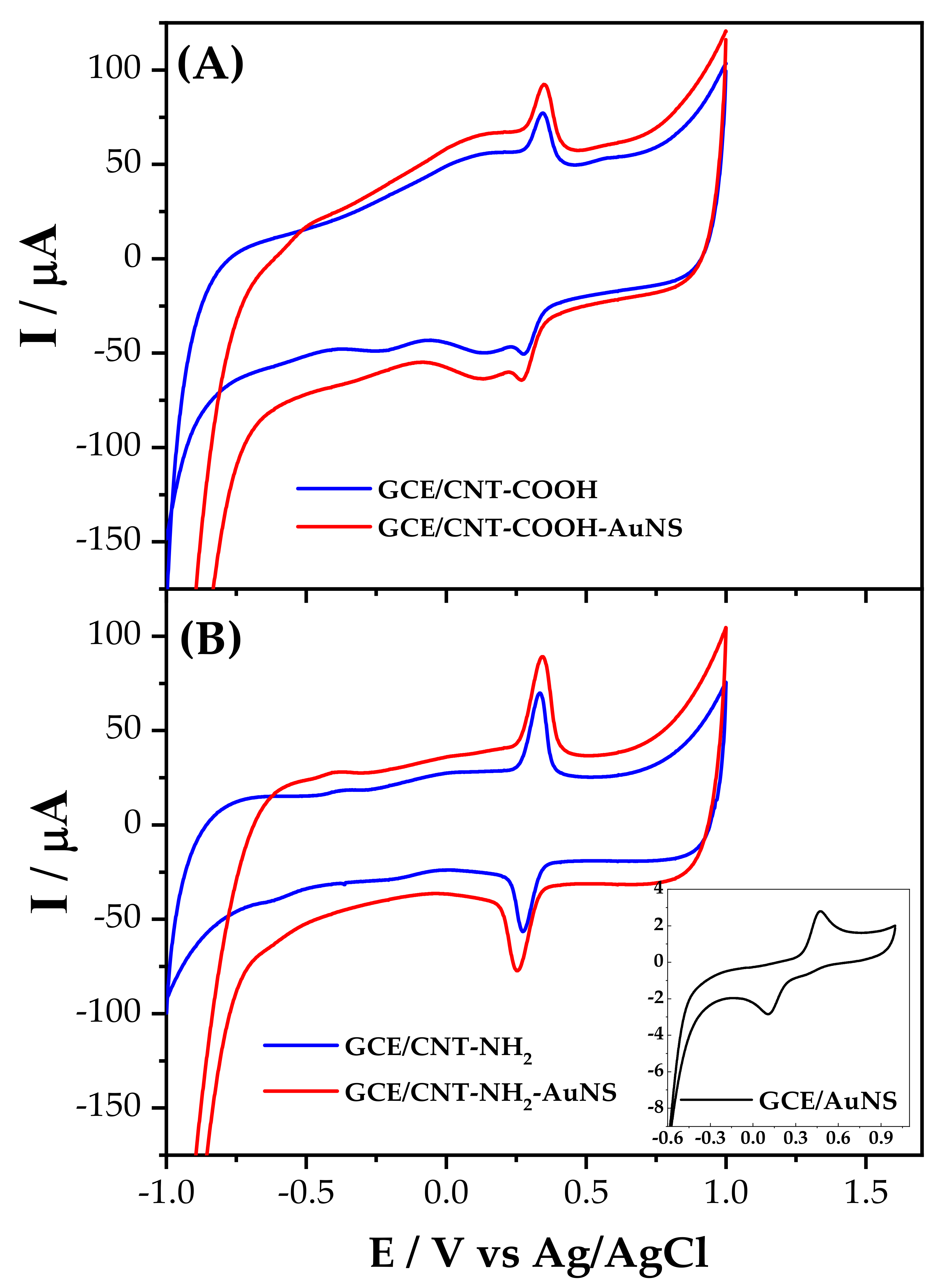
3.3. Determination of HQ Using GCE Modified with Different MWCNTs and AuNP
3.4. Simultaneous Determination of HQ and CT Using MWCNT/NH2-AuNP
3.5. Reproducibility Study of the Modified Electrode Area with Nanostructured Material
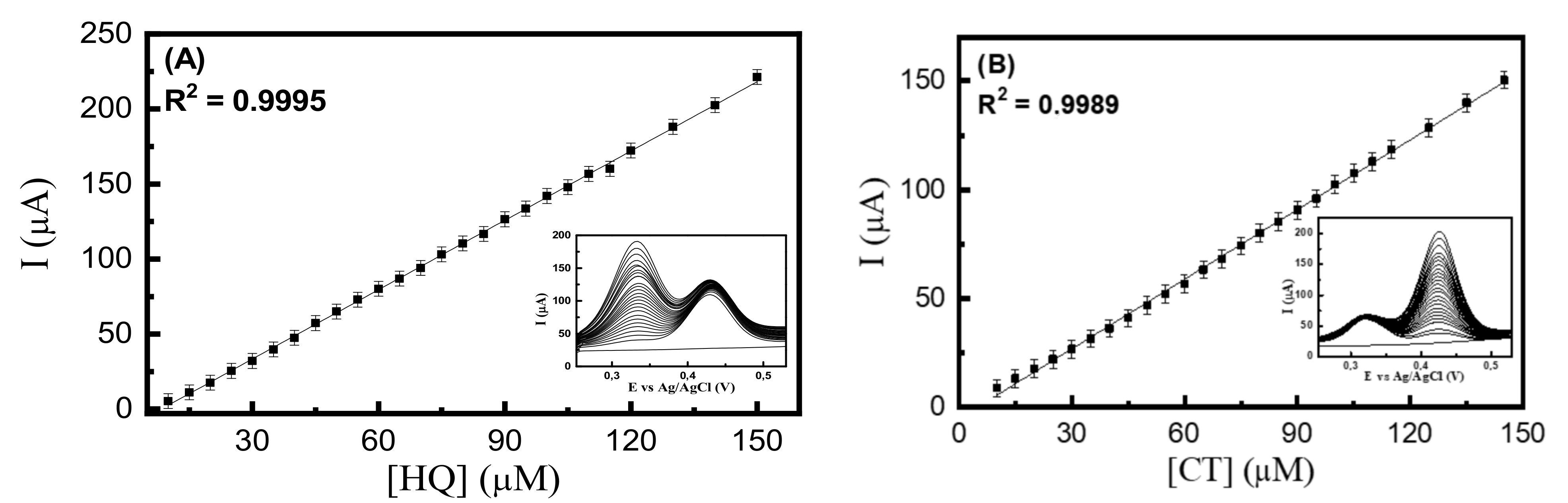
3.6. Determination of HQ and CT in Different Aqueous Matrices
3.7. Evaluation of Multivariate Calibration Method for Simultaneous Determination of HQ and CT
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Bin Zheng, J. Comparative investigation on electrochemical behavior of hydroquinone at carbon ionic liquid electrode, ionic liquid modified carbon paste electrode and carbon paste electrode. Electrochim. Acta 2007, 52, 7210–7216. [Google Scholar] [CrossRef]
- Vilela, C.L.S.; Bassin, J.P.; Peixoto, R.S. Water contamination by endocrine disruptors: Impacts, microbiological aspects and trends for environmental protection. Environ. Pollut. 2018, 235, 546–559. [Google Scholar] [CrossRef]
- Liu, W.; Tao, F.; Zhang, W.; Li, S.; Zheng, M. Contamination and emission factors of PCDD/Fs, unintentional PCBs, HxCBz, PeCBz and polychlorophenols in chloranil in China. Chemosphere 2012, 86, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Gmurek, M.; Olak-Kucharczyk, M.; Ledakowicz, S. Photochemical decomposition of endocrine disrupting compounds—A review. Chem. Eng. J. 2017, 310, 437–456. [Google Scholar] [CrossRef]
- Omar, T.F.T.; Ahmad, A.; Aris, A.Z.; Yusoff, F.M. Endocrine Disrupting Compounds (EDCs) in environmental matrices: Review of analytical strategies for pharmaceuticals, estrogenic hormones, and alkylphenol compounds. TrAC Trends Anal. Chem. 2016, 85, 241–259. [Google Scholar] [CrossRef]
- Aragó, M.; Ariño, C.; Dago, À.; Díaz-Cruz, J.M.; Esteban, M. Simultaneous determination of hydroquinone, catechol and resorcinol by voltammetry using graphene screen-printed electrodes and partial least squares calibration. Talanta 2016, 160, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Lawal, A.T. Progress in utilisation of graphene for electrochemical biosensors. Biosens. Bioelectron. 2018, 106, 149–178. [Google Scholar] [CrossRef]
- Qin, L.; Zeng, G.; Lai, C.; Huang, D.; Xu, P.; Zhang, C.; Cheng, M.; Liu, X.; Liu, S.; Li, B.; et al. “Gold rush” in modern science: Fabrication strategies and typical advanced applications of gold nanoparticles in sensing. Coord. Chem. Rev. 2018, 359, 1–31. [Google Scholar] [CrossRef]
- Gupta, S.; Murthy, C.; Prabha, C.R. Recent advances in carbon nanotube based electrochemical biosensors. Int. J. Biol. Macromol. 2018, 108, 687–703. [Google Scholar] [CrossRef]
- Rashidiani, J.; Kamali, M.; Sedighian, H.; Akbariqomi, M.; Mansouri, M.; Kooshki, H. Ultrahigh sensitive enhanced-electrochemiluminescence detection of cancer biomarkers using silica NPs/graphene oxide: A comparative study. Biosens. Bioelectron. 2018, 102, 226–233. [Google Scholar] [CrossRef]
- Govindasamy, M.; Manavalan, S.; Chen, S.-M.; Rajaji, U.; Chen, T.-W.; Al-Hemaid, F.M.A.; Ali, M.A.; Elshikh, M.S. Determination of neurotransmitter in biological and drug samples using gold nanorods decoratedf-MWCNTs modified electrode. J. Electrochem. Soc. 2018, 165, B370–B377. [Google Scholar] [CrossRef]
- Peng, C.; Li, Z.; Zhang, X.; Zhou, S.; Zhang, W.; Liu, X.; Zhao, P. Simultaneous determination of hydroquinone, catechol and resorcinol with high selectivity based on hollow nitrogen-doped mesoporous carbon spheres decorated graphene. J. Electrochem. Soc. 2018, 165, B212–B219. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Liu, H.; Chen, S.; Guo, X.; Lin, M.; Li, F. A portable electrochemical platform integrated with a 3D AuNPs/CNTs sponge for point-of-care testing of neurotransmitters. J. Electrochem. Soc. 2019, 166, B524–B531. [Google Scholar] [CrossRef]
- Wang, J. Carbon-nanotube based electrochemical biosensors: A review. Electroanalysis 2005, 17, 7–14. [Google Scholar] [CrossRef]
- Sedghi, R.; Pezeshkian, Z. Fabrication of non-enzymatic glucose sensor based on nanocomposite of MWCNTs-COOH-Poly(2-aminothiophenol)-Au NPs. Sens. Actuators B Chem. 2015, 219, 119–124. [Google Scholar] [CrossRef]
- Żelechowska, K.; Trawiński, B.; Dramińska, S.; Majdecka, D.; Bilewicz, R.; Kusz, B. Oxygen biosensor based on carbon nanotubes directly grown on graphitic substrate. Sens. Actuators B Chem. 2017, 240, 1308–1313. [Google Scholar] [CrossRef]
- Wong, A.; Foguel, M.V.; Khan, S.; de Oliveira, F.M.; Tarley, C.R.T.; Sotomayor, M.D. Development of an electrochemical sensor modified with MWCNT-COOH and MIP for detection of diuron. Electrochim. Acta 2015, 182, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Bu, C.; Liu, X.; Zhang, Y.; Li, L.; Zhou, X.; Lu, X. A sensor based on the carbon nanotubes-ionic liquid composite for simultaneous determination of hydroquinone and catechol. Colloids Surf. B Biointerfaces 2011, 88, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Ghoreishi, S.M.; Behpour, M.; Hajisadeghian, E.; Golestaneh, M. Voltammetric determination of resorcinol on the surface of a glassy carbon electrode modified with multi-walled carbon nanotube. Arab. J. Chem. 2016, 9, S1563–S1568. [Google Scholar] [CrossRef] [Green Version]
- Nugent, J.M.; Santhanam, K.S.V.; Rubio, A.; Ajayan, P.M. Fast electron transfer kinetics on multiwalled carbon nanotube microbundle electrodes. Nano Lett. 2001, 1, 87–91. [Google Scholar] [CrossRef]
- Liu, X.; Ding, Z.; He, Y.; Xue, Z.; Zhao, X.; Lu, X. Electrochemical behavior of hydroquinone at multi-walled carbon nanotubes and ionic liquid composite film modified electrode. Colloids Surf. B Biointerfaces 2010, 79, 27–32. [Google Scholar] [CrossRef]
- Mendes, R.G.; Wróbel, P.S.; Bachmatiuk, A.; Sun, J.; Gemming, T.; Liu, Z.; Rümmeli, M.H. Carbon nanostructures as a multi-functional platform for sensing applications. Chemosensors 2018, 6, 60. [Google Scholar] [CrossRef] [Green Version]
- Campbell, F.W.; Compton, R.G. The use of nanoparticles in electroanalysis: An updated review. Anal. Bioanal. Chem. 2009, 396, 241–259. [Google Scholar] [CrossRef]
- Katz, E.; Willner, I.; Wang, J. Electroanalytical and bioelectroanalytical systems based on metal and semiconductor nanoparticles. Electroanalysis 2004, 16, 19–44. [Google Scholar] [CrossRef]
- Sebarchievici, I.; Taranu, B.O.; Birdeanu, M.; Rus, S.F.; Fagadar-Cosma, E. Electrocatalytic behaviour and application of manganese porphyrin/gold nanoparticle- surface modified glassy carbon electrodes. Appl. Surf. Sci. 2016, 390, 131–140. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Xia, H.; Bai, S.; Hartmann, J.; Wang, D. Synthesis of monodisperse quasi-spherical gold nanoparticles in water via silver(I)-assisted citrate reduction. Langmuir 2010, 26, 3585–3589. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Maduraiveeran, G. Electrochemical detection of chemical pollutants based on gold nanomaterials. Trends Environ. Anal. Chem. 2017, 14, 28–36. [Google Scholar] [CrossRef]
- Petrović, S.; Guzsvány, V.; Ranković, N.; Beljin, J.; Rončević, S.; Dalmacija, B.; Ashrafi, A.M.; Kónya, Z.; Švancara, I.; Vytřas, K. Trace level voltammetric determination of Zn(II) in selected nutrition related samples by bismuth-oxychloride-multiwalled carbon nanotube composite based electrode. Microchem. J. 2019, 146, 178–186. [Google Scholar] [CrossRef]
- Bounegru, A.V.; Apetrei, C. Voltammetric sensors based on nanomaterials for detection of caffeic acid in food supplements. Chemosensors 2020, 8, 41. [Google Scholar] [CrossRef]
- Cui, H.; He, C.X.; Zhao, G.W. Determination of polyphenols by HPLC with inhibited chemiluminescence detection. J. Chromatogr. A 1999, 855, 171–179. [Google Scholar] [CrossRef]
- Moldoveanu, S.C.; Kiser, M. Gas chromatography/mass spectrometry versus liquid chromatography/fluorescence detection in the analysis of phenols in mainstream cigarette smoke. J. Chromatogr. A 2007, 1141, 90–97. [Google Scholar] [CrossRef]
- Elghobashy, M.R.; Bebawy, L.I.; Shokry, R.F.; Abbas, S.S. Successive ratio subtraction coupled with constant multiplication spectrophotometric method for determination of hydroquinone in complex mixture with its degradation products, tretinoin and methyl paraben. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 157, 116–123. [Google Scholar] [CrossRef]
- Patil, S.K.; Patil, S.A.; Vadiyar, M.M.; Awale, D.V.; Sartape, A.S.; Walekar, L.S.; Kolekar, G.B.; Ghorpade, U.V.; Kim, J.H.; Kolekar, S.S. Tailor-made dicationic ionic liquid as a fluorescent sensor for detection of hydroquinone and catechol. J. Mol. Liq. 2017, 244, 39–45. [Google Scholar] [CrossRef]
- Karim-Nezhad, G.; Moghaddam, M.H.; Khorablou, Z.; Dorraji, P.S. L-cysteine based polymer matrix decorated with au-nanoparticles: As a sensing platform for simultaneous determination of hydroquinone and catechol. J. Electrochem. Soc. 2017, 164, B193–B199. [Google Scholar] [CrossRef]
- Ahammad, A.J.S.; Akter, T.; Mamun, A.; Islam, T.; Hasan, M.; Mamun, M.A.; Faraezi, S.; Monira, F.Z.; Saha, J.K. Cost-effective electrochemical sensor based on carbon nanotube modified-pencil electrode for the simultaneous determination of hydroquinone and catechol. J. Electrochem. Soc. 2018, 165, B390–B397. [Google Scholar] [CrossRef]
- Otto, M. Chemometrics: Statics and Computer Application in Analytical Chemistry, 3rd ed.; Wiley: Weinheim, Germany, 2017; ISBN 9783527340972. [Google Scholar]
- Kumar, N.; Bansal, A.; Sarma, G.; Rawal, R.K. Chemometrics tools used in analytical chemistry: An overview. Talanta 2014, 123, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Li, B.; Luo, X. Carbon nanotube doped poly(3,4-ethylenedioxythiophene) for the electrocatalytic oxidation and detection of hydroquinone. Sens. Actuators B Chem. 2013, 176, 69–74. [Google Scholar] [CrossRef]
- Liu, Y.; Liao, H.; Zhou, Y.; Du, Y.; Wei, C.; Zhao, J.; Sun, S.; Loo, J.S.; Xu, Z.J. Fe2O3 nanoparticle/SWCNT composite electrode for sensitive electrocatalytic oxidation of hydroquinone. Electrochim. Acta 2015, 180, 1059–1067. [Google Scholar] [CrossRef]
- Zhang, X.P.; Jiang, W.L.; Cao, S.H.; Sun, H.J.; You, X.Q.; Cai, S.H.; Wang, J.L.; Zhao, C.S.; Wang, X.; Chen, Z.; et al. NMR spectroelectrochemistry in studies of hydroquinone oxidation by polyaniline thin films. Electrochim. Acta 2018, 273, 300–306. [Google Scholar] [CrossRef]
- Naqvi, S.T.R.; Rasheed, T.; Hussain, D.; Haq, M.N.U.; Majeed, S.; Shafi, S.; Ahmed, N.; Nawaz, R. Modification strategies for improving the solubility/dispersion of carbon nanotubes. J. Mol. Liq. 2020, 297, 111919. [Google Scholar] [CrossRef]
- Hu, Y.; Li, D.; Tang, P.; Bin, Y.; Wang, H. Comparative study of structure, mechanical and electromagnetic interference shielding properties of carbon nanotube buckypapers prepared by different dispersion media. Mater. Des. 2019, 184, 108175. [Google Scholar] [CrossRef]
- Cañete-Rosales, P.; Ortega, V.; Álvarez-Lueje, A.; Bollo, S.; González, M.; Ansón, A.; Martínez, M.T. Influence of size and oxidative treatments of multi-walled carbon nanotubes on their electrocatalytic properties. Electrochim. Acta 2012, 62, 163–171. [Google Scholar] [CrossRef]
- Ganesh, P.; Swamy, B.K. Simultaneous electroanalysis of hydroquinone and catechol at poly(brilliant blue) modified carbon paste electrode: A voltammetric study. J. Electroanal. Chem. 2015, 756, 193–200. [Google Scholar] [CrossRef]
- Tserpes, K.; Silvestre, N. Modeling of Carbon Nanotubes, Graphene and Their Composites; Springer International Publishing: Berlin/Heidelberg, Germany, 2014; ISBN 9783319012001. [Google Scholar]
- Zhang, Y.; Kang, Z. Highly conductive and anticorrosion Ag/CNTs/NDs hybrid films on molecular-grafted PET substrate for flexible electrodes. Appl. Surf. Sci. 2018, 427, 282–292. [Google Scholar] [CrossRef]
- Richard, G.C.; Batchelor-McAuley, C.; Dickinson, E.J.F. Understanding Voltammetry: Problems and Solutions; Imperial College Press: London, UK, 2012; ISBN 9781848167308. [Google Scholar]
- Yang, C.; Denno, M.E.; Pyakurel, P.; Venton, B.J. Recent trends in carbon nanomaterial-based electrochemical sensors for biomolecules: A review. Anal. Chim. Acta 2015, 887, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Alim, S.; Vejayan, J.; Yusoff, M.M.; Kafi, A. Recent uses of carbon nanotubes & gold nanoparticles in electrochemistry with application in biosensing: A review. Biosens. Bioelectron. 2018, 121, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, A.C. Practical guidelines for reporting results in single- and multi-component analytical calibration: A tutorial. Anal. Chim. Acta 2015, 868, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.N. Basic statistical methods for analytical chemistry. Part 2. Calibration and regression methods. A review. Analyst 1991, 116, 3–14. [Google Scholar] [CrossRef]
- Zheng, X.; Fan, R.; Hu, Y.; Zhong, H.; Yang, X.; Lv, R.; Yang, X.; Huang, B. Selective and simultaneous determination of hydroquinone and catechol by using a nitrogen-doped bagasse activated carbon modified electrode. Mater. Chem. Phys. 2020, 242, 122525. [Google Scholar] [CrossRef]
- Wang, J.; Yang, J.; Xu, P.; Liu, H.; Zhang, L.; Zhang, S.; Tian, L. Gold nanoparticles decorated biochar modified electrode for the high-performance simultaneous determination of hydroquinone and catechol. Sens. Actuators B Chem. 2020, 306, 127590. [Google Scholar] [CrossRef]
- Tashkhourian, J.; Daneshi, M.; Nami-Ana, F.; Behbahani, M.; Bagheri, A. Simultaneous determination of hydroquinone and catechol at gold nanoparticles mesoporous silica modified carbon paste electrode. J. Hazard. Mater. 2016, 318, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; He, Z.; Lian, Q.; Li, Z.; Jiang, H.; Lu, X. Simultaneous determination of dihydroxybenzene isomers based on graphene-graphene oxide nanocomposite modified glassy carbon electrode. Sens. Actuators B Chem. 2014, 193, 198–204. [Google Scholar] [CrossRef]
- Zhao, L.; Yu, J.; Yue, S.; Zhang, L.; Wang, Z.; Guo, P.; Liu, Q. Nickel oxide/carbon nanotube nanocomposites prepared by atomic layer deposition for electrochemical sensing of hydroquinone and catechol. J. Electroanal. Chem. 2018, 808, 245–251. [Google Scholar] [CrossRef]
- Han, H.S.; You, J.-M.; Seol, H.; Jeong, H.; Jeon, S. Electrochemical sensor for hydroquinone and catechol based on electrochemically reduced GO–terthiophene–CNT. Sens. Actuators B Chem. 2014, 194, 460–469. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Analyte | Sensitivity | Lineal Range/µM | LD/µM | LQ/µM | Δ Ep (HQ-CC)/mV | Technique | pH | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Graphene screen-printed electrodes | HQ | 1.3221 µM | n.d. | 2.7 | 9.1 | 105 | DPV | 7.0 | [6] |
| CC | 1.5825 µM | n.d. | 1.7 | 5.6 | |||||
| NDSBAC 1 | HQ | 0.9997 µM | 0.5–300 | 0.11 | n.d. | 112 | DPV | 6.5 | [53] |
| CC | 1.0662 µM | 0.5–300 | 0.09 | n.d. | |||||
| WBC/Au-850-15 2 | HQ | 164.4 μA µM−1 cm−2 | 0.008–1 | 0.002 | n.d. | 112.8 | DPV | 6.0 | [54] |
| CC | 132.0 μA µM−1 cm−2 | 0.01–1.0 | 0.004 | n.d. | |||||
| AuNPs-MPS 3 | HQ | n.d. | 10.0–1000.0 | 1.2 | n.d. | 123 | SWV | 7.0 | [55] |
| CC | n.d. | 30.0–1000.0 | 1.1 | n.d. | |||||
| GR-GO | HQ | n.d. | 0.5–300 | 0.16 | n.d. | 102 | DPV | 7.0 | [56] |
| CC | n.d. | 0.5–300 | 0.2 | n.d. | |||||
| NiO/MWCNT | HQ | n.d. | 10–500 | 2.5 | n.d. | ~110 | DPV | 7.0 | [57] |
| CC | n.d. | 10–400 | 2.5 | n.d. | |||||
| GO–TT–MWCNT | HQ | n.d. | 0.01–200 | 0.035 | n.d. | n.d. | DPV | 7.4 | [58] |
| CC | n.d. | 0.5–200 | 0.049 | n.d. | |||||
| MWCNT-NH2-AuNP | HQ | 1.539 µA µM−1 | 4.3–150.0 | 1.28 | 4.25 | 100 | DPV | 2.0 | This work |
| CC | 1.067 µA µM−1 | 3.9–150.0 | 1.06 | 3.87 |
| Matrix | Analyte | Concentration (µM) | Obtained Concentration (µM) | Recovery (%) |
|---|---|---|---|---|
| Drinking water | HQ | 55.0 | 48.6 ± 0.7 | 88.4 |
| CT | 45.3 ± 0.5 | 82.4 | ||
| Viticultural wastewater | HQ | 145.0 | 75.8 ± 0.8 | 52.3 |
| CT | 71.5 ± 0.4 | 49.3 | ||
| River water | HQ | 100.0 | 80.3 ± 0.5 | 80.3 |
| CT | 84.2 ± 0.6 | 84.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piña, S.; Candia-Onfray, C.; Hassan, N.; Jara-Ulloa, P.; Contreras, D.; Salazar, R. Glassy Carbon Electrode Modified with C/Au Nanostructured Materials for Simultaneous Determination of Hydroquinone and Catechol in Water Matrices. Chemosensors 2021, 9, 88. https://doi.org/10.3390/chemosensors9050088
Piña S, Candia-Onfray C, Hassan N, Jara-Ulloa P, Contreras D, Salazar R. Glassy Carbon Electrode Modified with C/Au Nanostructured Materials for Simultaneous Determination of Hydroquinone and Catechol in Water Matrices. Chemosensors. 2021; 9(5):88. https://doi.org/10.3390/chemosensors9050088
Chicago/Turabian StylePiña, Samuel, Christian Candia-Onfray, Natalia Hassan, Paola Jara-Ulloa, David Contreras, and Ricardo Salazar. 2021. "Glassy Carbon Electrode Modified with C/Au Nanostructured Materials for Simultaneous Determination of Hydroquinone and Catechol in Water Matrices" Chemosensors 9, no. 5: 88. https://doi.org/10.3390/chemosensors9050088
APA StylePiña, S., Candia-Onfray, C., Hassan, N., Jara-Ulloa, P., Contreras, D., & Salazar, R. (2021). Glassy Carbon Electrode Modified with C/Au Nanostructured Materials for Simultaneous Determination of Hydroquinone and Catechol in Water Matrices. Chemosensors, 9(5), 88. https://doi.org/10.3390/chemosensors9050088