
Mucosal Immune System Dysregulation in the Pathogenesis of IgA Nephropathy

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
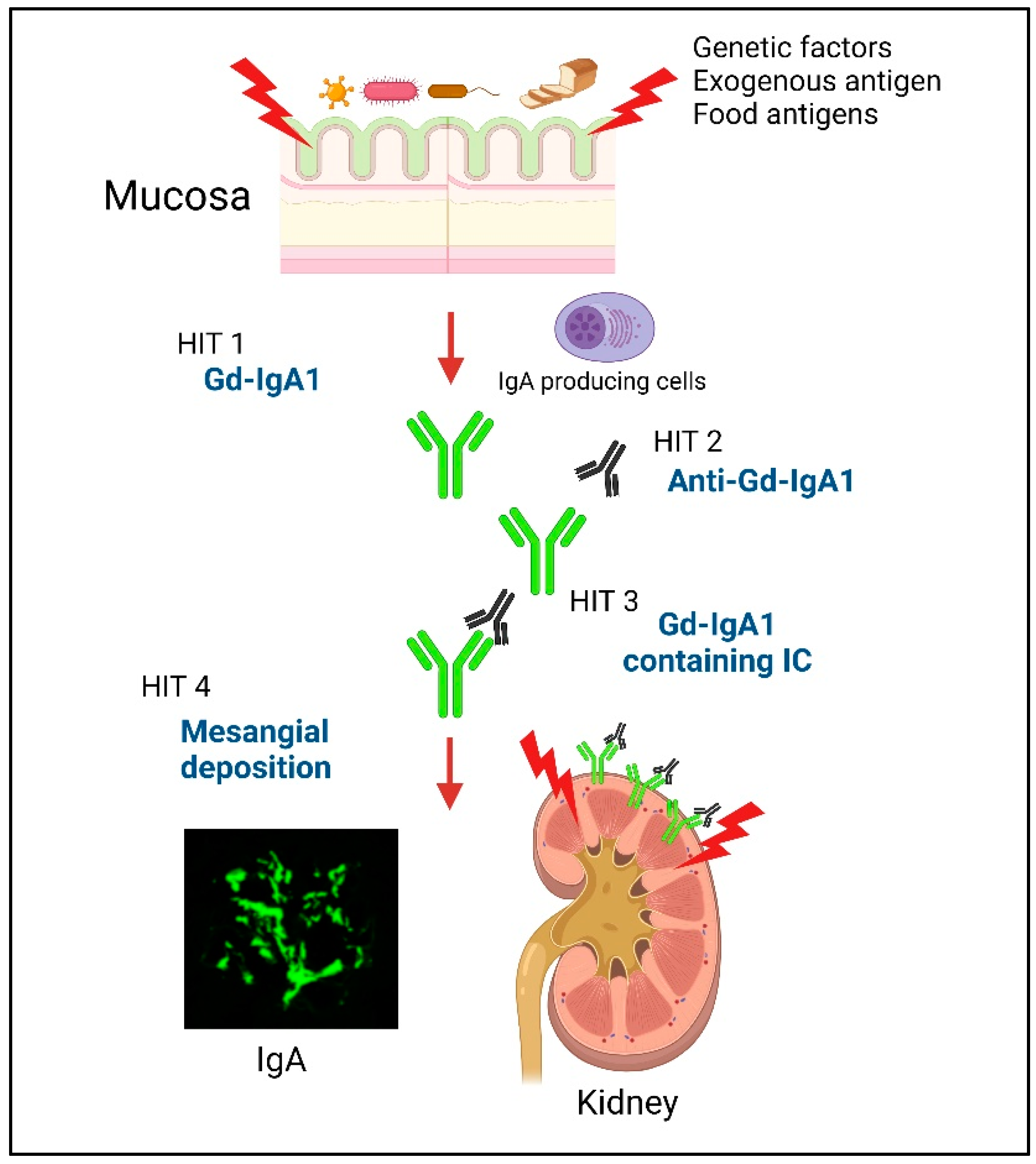
2. Multi-Hit Mechanism of IgAN
3. The Main Production Site of Gd-IgA1 in IgAN
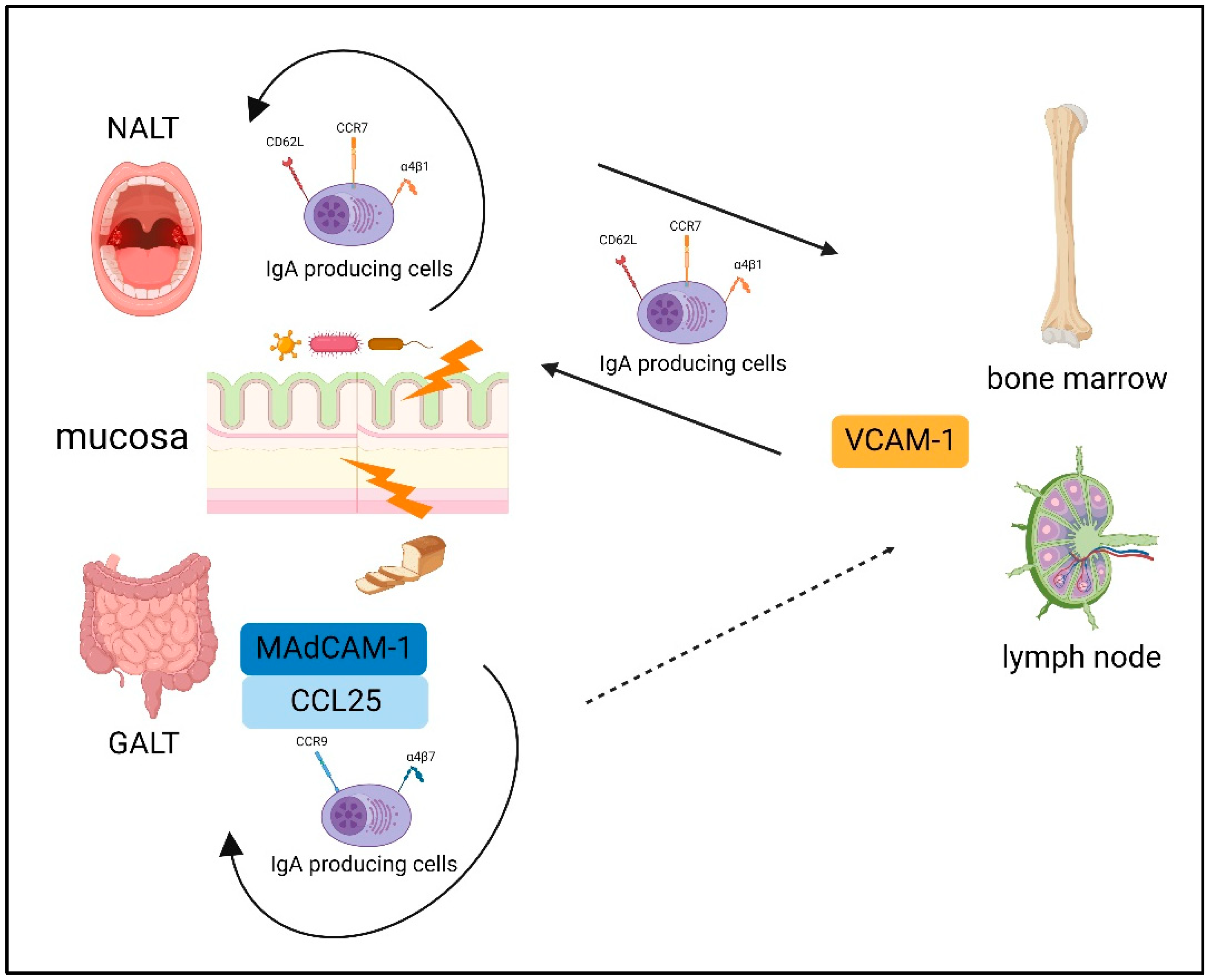
4. Gut-Associated Lymphoid Tissue (GALT)
5. Nasal-Associated Lymphoid Tissue (NALT)
6. The Difference between the NALT and GALT
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Berger, J.; Hinglais, N. Intercapillary deposits of IgA-IgG. J. Urol. Nephrol. 1968, 74, 694–695. [Google Scholar]
- Strippoli, G.F.; Maione, A.; Schena, F.P.; Tognoni, G.; Craig, J.C. IgA nephropathy: A disease in search of a large-scale clinical trial to reliably inform practice. Am. J. Kidney Dis. 2009, 53, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barratt, J.; Feehally, J. IgA nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2088–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharavi, A.G.; Yan, Y.; Scolari, F.; Schena, F.P.; Frasca, G.M.; Ghiggeri, G.M.; Cooper, K.; Amoroso, A.; Viola, B.F.; Battini, G.; et al. IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23. Nat. Genet. 2000, 26, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Tomino, Y.; Sakai, H. Exacerbating factors in patients with IgA nephropathy. Semin. Nephrol. 1987, 7, 315–317. [Google Scholar]
- Xie, Y.; Chen, X.; Nishi, S.; Narita, I.; Gejyo, F. Relationship between tonsils and IgA nephropathy as well as indications of tonsillectomy. Kidney Int. 2004, 65, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Kano, T.; Suzuki, H.; Makita, Y.; Fukao, Y.; Suzuki, Y. Nasal-associated lymphoid tissue is the major induction site for nephritogenic IgA in murine IgA nephropathy. Kidney Int. 2021, 100, 364–376. [Google Scholar] [CrossRef]
- Hricik, D.E.; Chung-Park, M.; Sedor, J.R. Glomerulonephritis. N. Engl. J. Med. 1998, 339, 888–899. [Google Scholar] [CrossRef]
- Pawar, R.D.; Patole, P.S.; Wornle, M.; Anders, H.J. Microbial nucleic acids pay a Toll in kidney disease. Am. J. Physiol. Ren. Physiol. 2006, 291, F509–F516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyono, H.; Fukuyama, S. NALT- versus Peyer’s-patch-mediated mucosal immunity. Nat. Rev. Immunol. 2004, 4, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Yuki, Y.; Kiyono, H. New generation of mucosal adjuvants for the induction of protective immunity. Rev. Med. Virol. 2003, 13, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.C.; Bailey, E.M.; Brenchley, P.E.; Buck, K.S.; Barratt, J.; Feehally, J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001, 60, 969–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiki, Y.; Odani, H.; Takahashi, M.; Yasuda, Y.; Nishimoto, A.; Iwase, H.; Shinzato, T.; Kobayashi, Y.; Maeda, K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001, 59, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasutake, J.; Suzuki, Y.; Suzuki, H.; Hiura, N.; Yanagawa, H.; Makita, Y.; Kaneko, E.; Tomino, Y. Novel lectin-independent approach to detect galactose-deficient IgA1 in IgA nephropathy. Nephrol. Dial. Transplant. 2015, 30, 1315–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baenziger, J.; Kornfeld, S. Structure of the Carbohydrate Units of IgA1 Immunoglobulin. J. Biol. Chem. 1974, 249, 7270–7281. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Julian, B.A.; Tomana, M.; Mesteck, J. Progress in molecular and genetic studies of IgA nephropathy. J. Clin. Immunol. 2001, 21, 310–327. [Google Scholar] [CrossRef]
- Piller, V.; Piller, F.; Fukuda, M. Biosynthesis of truncated O-glycans in the T cell line Jurkat. Localization of O-glycan initiation. J. Biol. Chem. 1990, 265, 9264–9271. [Google Scholar] [CrossRef]
- Suzuki, H.; Moldoveanu, Z.; Hall, S.; Brown, R.; Vu, H.L.; Novak, L.; Julian, B.A.; Tomana, M.; Wyatt, R.J.; Edberg, J.C.; et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J. Clin. Investig. 2008, 118, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Allegri, L.; Suzuki, Y.; Hall, S.; Moldoveanu, Z.; Wyatt, R.J.; Novak, J.; Julian, B.A. Galactose-Deficient IgA1 as a Candidate Urinary Polypeptide Marker of IgA Nephropathy? Dis. Markers 2016, 2016, 7806438. [Google Scholar] [CrossRef] [Green Version]
- Fukao, Y.; Suzuki, H.; Kim, J.S.; Jeong, K.H.; Makita, Y.; Kano, T.; Nihei, Y.; Nakayama, M.; Lee, M.; Kato, R.; et al. Galactose-Deficient IgA1 as a Candidate Urinary Marker of IgA Nephropathy. J. Clin. Med. 2022, 11, 3173. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, Z.; Wyatt, R.J.; Lee, J.Y.; Tomana, M.; Julian, B.A.; Mestecky, J.; Mestecky, J.; Huang, W.-Q.; Anreddy, S.R.; Hall, S.; et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007, 71, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Gharavi, A.G.; Moldoveanu, Z.; Wyatt, R.J.; Barker, C.V.; Woodford, S.Y.; Lifton, R.P.; Mestecky, J.; Novak, J.; Julian, B.A. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Matsuzaki, K.; Suzuki, H.; Okazaki, K.; Yanagawa, H.; Ieiri, N.; Sato, M.; Sato, T.; Taguma, T.; Matsuoka, J.; et al. Serum levels of galactose-deficient immunoglobulin (Ig) A1 and related immune complex are associated with disease activity of IgA nephropathy. Clin. Exp. Nephrol. 2014, 18, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Yanagihara, T.; Brown, R.; Hall, S.; Moldoveanu, Z.; Goepfert, A.; Tomana, M.; Julian, B.A.; Mestecky, J.; Novak, J. In vitro-generated immune complexes containing galactose-deficient IgA1 stimulate proliferation of mesangial cells. Results Immunol. 2012, 2, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomana, M.; Novak, J.; Julian, B.A.; Matousovic, K.; Konecny, K.; Mestecky, J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J. Clin. Investig. 1999, 104, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Fan, R.; Zhang, Z.; Brown, R.; Hall, S.; Julian, B.A.; Chatham, W.W.; Suzuki, Y.; Wyatt, R.J.; Moldoveanu, Z.; et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Investig. 2009, 119, 1668–1677. [Google Scholar] [CrossRef] [Green Version]
- Rizk, D.V.; Saha, M.K.; Hall, S.; Novak, L.; Brown, R.; Huang, Z.Q.; Fatima, H.; Julian, B.A.; Novak, J. Glomerular Immunodeposits of Patients with IgA Nephropathy Are Enriched for IgG Autoantibodies Specific for Galactose-Deficient IgA1. J. Am. Soc. Nephrol. 2019, 30, 2017–2026. [Google Scholar] [CrossRef]
- Yamaji, K.; Suzuki, Y.; Suzuki, H.; Satake, K.; Horikoshi, S.; Novak, J.; Tomino, Y. The kinetics of glomerular deposition of nephritogenic IgA. PLoS ONE 2014, 9, e113005. [Google Scholar] [CrossRef] [Green Version]
- Makita, Y.; Suzuki, H.; Nakano, D.; Yanagawa, H.; Kano, T.; Novak, J.; Nishiyama, A.; Suzuki, Y. Glomerular deposition of galactose-deficient IgA1-containing immune complexes via glomerular endothelial cell injuries. Nephrol. Dial. Transplant. 2022, 37, 1629–1636. [Google Scholar] [CrossRef]
- Wyatt, R.J.; Julian, B.A. IgA nephropathy. N. Engl. J. Med. 2013, 368, 2402–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oortwijn, B.D.; Rastaldi, M.P.; Roos, A.; Mattinzoli, D.; Daha, M.R.; van Kooten, C. Demonstration of secretory IgA in kidneys of patients with IgA nephropathy. Nephrol. Dial. Transplant. 2007, 22, 3191–3195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Xu, L.X.; Liu, G.; Zhao, M.H.; Wang, H.Y. The level of serum secretory IgA of patients with IgA nephropathy is elevated and associated with pathological phenotypes. Nephrol. Dial. Transplant. 2008, 23, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandtzaeg, P.; Johansen, F.E. Mucosal B cells: Phenotypic characteristics, transcriptional regulation, and homing properties. Immunol. Rev. 2005, 206, 32–63. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P.; Baekkevold, E.S.; Farstad, I.N.; Jahnsen, F.L.; Johansen, F.E.; Nilsen, E.M.; Yamanaka, T. Regional specialization in the mucosal immune system: What happens in the microcompartments? Immunol. Today 1999, 20, 141–151. [Google Scholar] [CrossRef]
- Sakai, O. IgA nephropathy: Current concept and feature trends. Nephrology 1997, 3, 2–3. [Google Scholar]
- Imasawa, T.; Utsunomiya, Y. Stem cells in renal biology: Bone marrow transplantation for the treatment of IgA nephropathy. Exp. Nephrol. 2002, 10, 51–58. [Google Scholar] [CrossRef]
- Suzuki, H.; Suzuki, Y.; Aizawa, M.; Yamanaka, T.; Kihara, M.; Pang, H.; Horikoshi, S.; Tomino, Y. Th1 polarization in murine IgA nephropathy directed by bone marrow-derived cells. Kidney Int. 2007, 72, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Tomino, Y. The mucosa-bone-marrow axis in IgA nephropathy. Contrib. Nephrol. 2007, 157, 70–79. [Google Scholar]
- Helin, H.; Mustonen, J.; Reunala, T.; Pasternack, A. IgA nephropathy associated with celiac disease and dermatitis herpetiformis. Arch. Pathol. Lab. Med. 1983, 107, 324–327. [Google Scholar]
- Collin, P.; Syrjanen, J.; Partanen, J.; Pasternack, A.; Kaukinen, K.; Mustonen, J. Celiac disease and HLA DQ in patients with IgA nephropathy. Am. J. Gastroenterol. 2002, 97, 2572–2576. [Google Scholar] [CrossRef]
- Coppo, R.; Basolo, B.; Rollino, C.; Roccatello, D.; Martina, G.; Amore, A.; Piccoli, G. Dietary gluten and primary IgA nephropathy. N. Engl. J. Med. 1986, 315, 1167–1168. [Google Scholar] [PubMed]
- Papista, C.; Lechner, S.; Ben Mkaddem, S.; LeStang, M.B.; Abbad, L.; Bex-Coudrat, J.; Pillebout, E.; Chemouny, J.M.; Jablonski, M.; Flamant, M.; et al. Gluten exacerbates IgA nephropathy in humanized mice through gliadin-CD89 interaction. Kidney Int. 2015, 88, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Emancipator, S.N.; Gallo, G.R.; Lamm, M.E. Experimental IgA nephropathy induced by oral immunization. J. Exp. Med. 1983, 157, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Li, Y.; Scolari, F.; Sanna-Cherchi, S.; Choi, M.; Verbitsky, M.; Fasel, D.; Lata, S.; Prakash, S.; Shapiro, S.; et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellström, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017, 389, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Coppo, R.; Mariat, C. Systemic corticosteroids and mucosal-associated lymphoid tissue-targeted therapy in immunoglobulin A nephropathy: Insight from the NEFIGAN study. Nephrol. Dial. Transplant. 2020, 35, 1291–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahne, M.; Kataoka, T.; Schroter, M.; Hofmann, K.; Irmler, M.; Bodmer, J.L.; Schneider, P.; Bornand, T.; Holler, N.; French, L.R.; et al. APRIL, a new ligand of the tumor necrosis factor family, stimulates tumor cell growth. J. Exp. Med. 1998, 188, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.D.; Kujawa, J.; Wilson, C.; Papandile, A.; Poreci, U.; Porfilio, E.A.; Ward, L.; Lawson, M.A.E.; Macpherson, A.J.; McCoy, K.D.; et al. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J. Clin. Investig. 2011, 121, 3991–4002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandtzaeg, P. Transport models for secretory IgA and secretory IgM. Clin. Exp. Immunol. 1981, 44, 221–232. [Google Scholar] [PubMed]
- Wang, J.; Anders, R.A.; Wu, Q.; Peng, D.; Cho, J.H.; Sun, Y.; Karaliukas, R.; Kang, H.-S.; Turner, J.R.; Fu, Y.-X. Dysregulated LIGHT expression on T cells mediates intestinal inflammation and contributes to IgA nephropathy. J. Clin. Investig. 2004, 113, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Arsenescu, R.; Bruno, M.E.; Rogier, E.W.; Stefka, A.T.; McMahan, A.E.; Wright, T.B.; Nasser, M.S.; Villiers, W.J.S.; Kaetzel, C.S. Signature biomarkers in Crohn’s disease: Toward a molecular classification. Mucosal. Immunol. 2008, 1, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Aizawa, M.; Suzuki, Y.; Suzuki, H.; Pang, H.; Kihara, M.; Nakata, J.; Yamaji, K.; Horikoshi, S.; Tomino, Y. Uncoupling of glomerular IgA deposition and disease progression in alymphoplasia mice with IgA nephropathy. PLoS ONE 2014, 9, e95365. [Google Scholar] [CrossRef]
- Moeller, S.; Canetta, P.A.; Taylor, A.K.; Arguelles-Grande, C.; Snyder, H.; Green, P.H.; Kiryluk, K.; Alaedini, A. Lack of serologic evidence to link IgA nephropathy with celiac disease or immune reactivity to gluten. PLoS ONE 2014, 9, e94677. [Google Scholar] [CrossRef] [PubMed]
- McKnight, A.J.; O’Donoghue, D.; Peter Maxwell, A. Annotated chromosome maps for renal disease. Hum. Mutat. 2009, 30, 314–320. [Google Scholar] [CrossRef]
- Kloster Smerud, H.; Fellstrom, B.; Hallgren, R.; Osagie, S.; Venge, P.; Kristjansson, G. Gastrointestinal sensitivity to soy and milk proteins in patients with IgA nephropathy. Clin. Nephrol. 2010, 74, 364–371. [Google Scholar] [CrossRef]
- Coppo, R.; Amore, A.; Roccatello, D. Dietary antigens and primary immunoglobulin A nephropathy. J. Am. Soc. Nephrol. 1992, 2 (Suppl. 10), S173–S180. [Google Scholar] [CrossRef]
- Komatsu, H.; Fujimoto, S.; Hara, S.; Sato, Y.; Yamada, K.; Kitamura, K. Effect of tonsillectomy plus steroid pulse therapy on clinical remission of IgA nephropathy: A controlled study. Clin. J. Am. Soc. Nephrol. 2008, 3, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Amano, H.; Kawamura, T.; Watanabe, K.; Koike, K.; Shimizu, A.; Endo, S.; Tsunoi, N.; Okonogi, H.; Niyazaki, Y.; et al. Tonsillectomy reduces recurrence of IgA nephropathy in mesangial hypercellularity type categorized by the Oxford classification. Clin. Exp. Nephrol. 2016, 20, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; He, L.; Peng, X.; Liu, H.; Peng, Y.; Yuan, S.; Liu, Y.; Chen, X.; Liu, F.; Liu, C. The efficacy of tonsillectomy on clinical remission and relapse in patients with IgA nephropathy: A randomized controlled trial. Ren. Fail. 2016, 38, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tang, Z.; Wang, Q.; Yu, Y.; Zeng, C.; Chen, H.; Liu, Z.H.; Li, L.S. Long-term efficacy of tonsillectomy in Chinese patients with IgA nephropathy. Am. J. Nephrol. 2007, 27, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Matsuzaki, K.; Yasuda, T.; Nishikawa, M.; Yasuda, Y.; Koike, K.; Maruyama, S.; Yokoo, T.; Matsuo, S.; Kawamura, T.; et al. Association Between Tonsillectomy and Outcomes in Patients with Immunoglobulin A Nephropathy. JAMA Netw. Open 2019, 2, e194772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Liu, D.; Duan, G.; Liu, Z. Long-term efficacy of tonsillectomy as a treatment in patients with IgA nephropathy: A meta-analysis. Int. Urol. Nephrol. 2017, 49, 103–112. [Google Scholar] [CrossRef]
- Kovacs, T.; Vas, T.; Kovesdy, C.P.; Degrell, P.; Nagy, G.; Rekasi, Z.; Wittmann, I.; Nagy, J. Effect of tonsillectomy and its timing on renal outcomes in Caucasian IgA nephropathy patients. Int. Urol. Nephrol. 2014, 46, 2175–2182. [Google Scholar] [CrossRef]
- Nagy, J.; Brandtzaeg, P. Tonsillar distribution of IgA and IgG immunocytes and production of IgA subclasses and J chain in tonsillitis vary with the presence or absence of IgA nephropathy. Scand. J. Immunol. 1988, 27, 393–399. [Google Scholar] [CrossRef]
- Bene, M.C.; Hurault De Ligny, B.; Kessler, M.; Faure, G.C. Confirmation of tonsillar anomalies in IgA nephropathy: A multicenter study. Nephron 1991, 58, 425–428. [Google Scholar] [CrossRef]
- Horie, A.; Hiki, Y.; Odani, H.; Yasuda, Y.; Takahashi, M.; Kato, M.; Iwase, H.; Kobayashi, Y.; Nakashima, I.; Maeda, K. IgA1 molecules produced by tonsillar lymphocytes are under-O-glycosylated in IgA nephropathy. Am. J. Kidney Dis. 2003, 42, 486–496. [Google Scholar] [CrossRef]
- Inoue, T.; Sugiyama, H.; Hiki, Y.; Takiue, K.; Morinaga, H.; Kitagawa, M.; Maeshima, Y.; Fukushima, K.; Nishizaki, K.; Akagi, H.; et al. Differential expression of glycogenes in tonsillar B lymphocytes in association with proteinuria and renal dysfunction in IgA nephropathy. Clin. Immunol. 2010, 136, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Kawabe, M.; Yamamoto, I.; Yamakawa, T.; Katsumata, H.; Isaka, N.; Katsuma, A.; Nakada, Y.; Kobayashi, A.; Koike, K.; Ueda, H.; et al. Association Between Galactose-Deficient IgA1 Derived from the Tonsils and Recurrence of IgA Nephropathy in Patients Who Underwent Kidney Transplantation. Front. Immunol. 2020, 11, 2068. [Google Scholar] [CrossRef]
- Suzuki, S.; Nakatomi, Y.; Sato, H.; Tsukada, H.; Arakawa, M. Haemophilus parainfluenzae antigen and antibody in renal biopsy samples and serum of patients with IgA nephropathy. Lancet 1994, 343, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Kai, H.; Shimizu, Y.; Hagiwara, M.; Yoh, K.; Hirayama, K.; Yamagata, K.; Ohba, S.; Nagata, M.; Koyama, A. Post-MRSA infection glomerulonephritis with marked Staphylococcus aureus cell envelope antigen deposition in glomeruli. J. Nephrol. 2006, 19, 215–219. [Google Scholar] [PubMed]
- Coppo, R.; Amore, A.; Peruzzi, L.; Vergano, L.; Camilla, R. Innate immunity and IgA nephropathy. J. Nephrol. 2010, 23, 626–632. [Google Scholar] [PubMed]
- Okazaki, K.; Suzuki, Y.; Otsuji, M.; Suzuki, H.; Kihara, M.; Kajiyama, T.; Hashimoto, A.; Nishimura, H.; Brown, R.; Hall, S.; et al. Development of a model of early-onset IgA nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1364–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Suzuki, Y.; Yamanaka, T.; Hirose, S.; Nishimura, H.; Toei, J.; Horikoshi, S.; Tomino, Y. Genome-wide scan in a novel IgA nephropathy model identifies a susceptibility locus on murine chromosome 10, in a region syntenic to human IGAN1 on chromosome 6q22-23. J. Am. Soc. Nephrol. 2005, 16, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Suzuki, Y.; Narita, I.; Aizawa, M.; Kihara, M.; Yamanaka, T.; Kanou, T.; Tsukaguchi, H.; Novak, J.; Horikoshi, S.; et al. Toll-like receptor 9 affects severity of IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2384–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, D.; Suzuki, Y.; Kano, T.; Suzuki, H.; Matsuoka, J.; Yokoi, H.; Horikoshi, S.; Ikeda, K.; Tomino, Y. Tonsillar TLR9 expression and efficacy of tonsillectomy with steroid pulse therapy in IgA nephropathy patients. Nephrol. Dial. Transplant. 2012, 27, 1090–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakata, J.; Suzuki, Y.; Suzuki, H.; Sato, D.; Kano, T.; Yanagawa, H.; Matsuzaki, K.; Horikoshi, S.; Novak, J.; Tomino, Y. Changes in nephritogenic serum galactose-deficient IgA1 in IgA nephropathy following tonsillectomy and steroid therapy. PLoS ONE 2014, 9, e89707. [Google Scholar] [CrossRef]
- Yu, X.Q.; Li, M.; Zhang, H.; Low, H.Q.; Wei, X.; Wang, J.Q.; Sun, L.D.; Sim, K.S.; Li, Y.; Foo, J.N.; et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 44, 178–182. [Google Scholar] [CrossRef]
- Stein, J.V.; López-Fraga, M.; Elustondo, F.A.; Carvalho-Pinto, C.E.; Rodríguez, D.; Gómez-Caro, R.; Jong, J.; Martinez-A, C.; Medema, J.P.; Hahne, M. APRIL modulates B and T cell immunity. J. Clin. Investig. 2002, 109, 1587–1598. [Google Scholar] [CrossRef]
- He, B.; Xu, W.; Santini, P.A.; Polydorides, A.D.; Chiu, A.; Estrella, J.; Shan, M.; Chadburn, A.; Villanacci, V.; Plebani, A.; et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 2007, 26, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Alvarez, M.; Suzuki, H.; Hirose, S.; Izui, S.; Tomino, Y.; Huard, B.; Suzuki, Y. Pathogenic Role of a Proliferation-Inducing Ligand (APRIL) in Murine IgA Nephropathy. PLoS ONE 2015, 10, e0137044. [Google Scholar] [CrossRef] [PubMed]
- Myette, J.R.; Kano, T.; Suzuki, H.; Sloan, S.E.; Szretter, K.J.; Ramakrishnan, B.; Adari, H.; Deotale, K.D.; Engler, F.; Shriver, Z.; et al. A Proliferation Inducing Ligand (APRIL) targeted antibody is a safe and effective treatment of murine IgA nephropathy. Kidney Int. 2019, 96, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Muto, M.; Manfroi, B.; Suzuki, H.; Joh, K.; Nagai, M.; Wakai, S.; Righini, C.; Maiguma, M.; Izui, S.; Tomino, Y.; et al. Toll-Like Receptor 9 Stimulation Induces Aberrant Expression of a Proliferation-Inducing Ligand by Tonsillar Germinal Center B Cells in IgA Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 1227–1238. [Google Scholar] [CrossRef] [Green Version]
- Goto, T.; Bandoh, N.; Yoshizaki, T.; Nozawa, H.; Takahara, M.; Ueda, S.; Hayashi, T.; Harabuchi, Y. Increase in B-cell-activation factor (BAFF) and IFN-gamma productions by tonsillar mononuclear cells stimulated with deoxycytidyl-deoxyguanosine oligodeoxynucleotides (CpG-ODN) in patients with IgA nephropathy. Clin. Immunol. 2008, 126, 260–269. [Google Scholar] [CrossRef]
- Li, W.; Peng, X.; Liu, Y.; Liu, H.; Liu, F.; He, L.; Liu, Y.; Zhang, F.; Guo, C.; Chen, G.; et al. TLR9 and BAFF: Their expression in patients with IgA nephropathy. Mol. Med. Rep. 2014, 10, 1469–1474. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Honda, K.; Shinkura, R.; Adachi, S.; Nishikawa, S.; Maki, K.; Ikuta, K.; Nishikawa, S. IL-7 receptor alpha+ CD3(-) cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int. Immunol. 1999, 11, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.E.; Yoshida, T.; Sime, W.; Hiepe, F.; Thiele, K.; Manz, R.A.; Radbruch, A.; Dorner, T. Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood 2009, 113, 2461–2469. [Google Scholar] [CrossRef]
- Rojas, O.L.; Probstel, A.K.; Porfilio, E.A.; Wang, A.A.; Charabati, M.; Sun, T.; Lee, D.S.W.; Galicia, G.; Ramaglia, V.; Ward, L.A.; et al. Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation via IL-10. Cell 2019, 176, 610–624 e18. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, A.J.; McCoy, K.D.; Johansen, F.E.; Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal. Immunol. 2008, 1, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Brandtzaeg, P. Secretory IgA: Designed for Anti-Microbial Defense. Front. Immunol. 2013, 4, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandtzaeg, P. Potential of nasopharynx-associated lymphoid tissue for vaccine responses in the airways. Am. J. Respir. Crit. Care Med. 2011, 183, 1595–1604. [Google Scholar] [CrossRef]
- Zachova, K.; Jemelkova, J.; Kosztyu, P.; Ohyama, Y.; Takahashi, K.; Zadrazil, J.; Orsag, J.; Matousovic, K.; Galuszkova, D.; Petejova, N.; et al. Galactose-Deficient IgA1 B cells in the Circulation of IgA Nephropathy Patients Carry Preferentially Lambda Light Chains and Mucosal Homing Receptors. J. Am. Soc. Nephrol. 2022, 33, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Monteiro, R.C.; Coppo, R.; Suzuki, H. The Phenotypic Difference of IgA Nephropathy and its Race/Gender-dependent Molecular Mechanisms. Kidney360 2021, 2, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Feehally, J.; Coppo, R.; Troyanov, S.; Bellur, S.S.; Cattran, D.; Cook, T.; Roberts, I.S.D.; Verhave, J.C.; Camilla, R.; Vergano, L.; et al. Tonsillectomy in a European Cohort of 1,147 Patients with IgA Nephropathy. Nephron 2016, 132, 15–24. [Google Scholar] [CrossRef]
- Piccoli, A.; Codognotto, M.; Tabbi, M.G.; Favaro, E.; Rossi, B. Influence of tonsillectomy on the progression of mesangioproliferative glomerulonephritis. Nephrol. Dial. Transplant. 2010, 25, 2583–2589. [Google Scholar] [CrossRef] [Green Version]
- Rasche, F.M.; Schwarz, A.; Keller, F. Tonsillectomy does not prevent a progressive course in IgA nephropathy. Clin. Nephrol. 1999, 51, 147–152. [Google Scholar]
- Kidney Disease: Improving Global Outcomes Glomerular Diseases Work G. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [CrossRef]
- Liu, L.L.; Wang, L.N.; Jiang, Y.; Yao, L.; Dong, L.P.; Li, Z.L.; Li, X.L. Tonsillectomy for IgA nephropathy: A meta-analysis. Am. J. Kidney Dis. 2015, 65, 80–87. [Google Scholar] [CrossRef]
- Taylor, S.; Pieri, K.; Nanni, P.; Tica, J.; Barratt, J.; Didangelos, A. Phosphatidylethanolamine binding protein-4 (PEBP4) is increased in IgA nephropathy and is associated with IgA-positive B-cells in affected kidneys. J. Autoimmun. 2019, 105, 102309. [Google Scholar] [CrossRef]
- Currie, E.G.; Coburn, B.; Porfilio, E.A.; Lam, P.; Rojas, O.L.; Novak, J.; Yang, S.; Chowdhury, R.B.; Ward, L.A.; Wang, P.W.; et al. Immunoglobulin A nephropathy is characterized by anticommensal humoral immune responses. JCI Insight 2022, 7, e141289. [Google Scholar] [CrossRef] [PubMed]
- McGrogan, A.; Franssen, C.F.; de Vries, C.S. The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol. Dial. Transplant. 2011, 26, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schena, F.P.; Nistor, I. Epidemiology of IgA Nephropathy: A Global Perspective. Semin. Nephrol. 2018, 38, 435–442. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kano, T.; Suzuki, H.; Makita, Y.; Nihei, Y.; Fukao, Y.; Nakayama, M.; Lee, M.; Kato, R.; Aoki, R.; Yamada, K.; et al. Mucosal Immune System Dysregulation in the Pathogenesis of IgA Nephropathy. Biomedicines 2022, 10, 3027. https://doi.org/10.3390/biomedicines10123027
Kano T, Suzuki H, Makita Y, Nihei Y, Fukao Y, Nakayama M, Lee M, Kato R, Aoki R, Yamada K, et al. Mucosal Immune System Dysregulation in the Pathogenesis of IgA Nephropathy. Biomedicines. 2022; 10(12):3027. https://doi.org/10.3390/biomedicines10123027
Chicago/Turabian StyleKano, Toshiki, Hitoshi Suzuki, Yuko Makita, Yoshihito Nihei, Yusuke Fukao, Maiko Nakayama, Mingfeng Lee, Rina Kato, Ryosuke Aoki, Koshi Yamada, and et al. 2022. "Mucosal Immune System Dysregulation in the Pathogenesis of IgA Nephropathy" Biomedicines 10, no. 12: 3027. https://doi.org/10.3390/biomedicines10123027
APA StyleKano, T., Suzuki, H., Makita, Y., Nihei, Y., Fukao, Y., Nakayama, M., Lee, M., Kato, R., Aoki, R., Yamada, K., Muto, M., & Suzuki, Y. (2022). Mucosal Immune System Dysregulation in the Pathogenesis of IgA Nephropathy. Biomedicines, 10(12), 3027. https://doi.org/10.3390/biomedicines10123027