
CK2 and the Hallmarks of Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Importance of CK2 in Different Hallmarks of Cancer
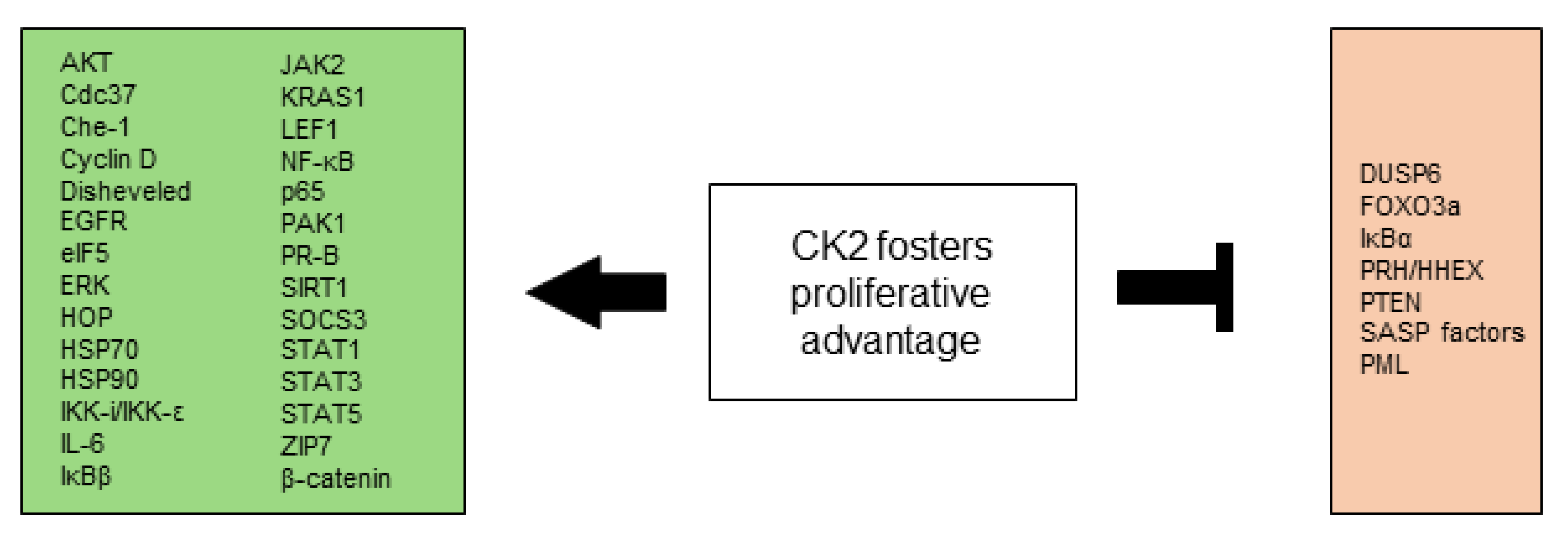
2.1. CK2 Is Involved in Selective Growth and Proliferative Advantage
2.1.1. Modulation of Signaling Pathways
2.1.2. Modulation of Transcription and Translation Factors
2.1.3. Modulation of Regulatory Proteins
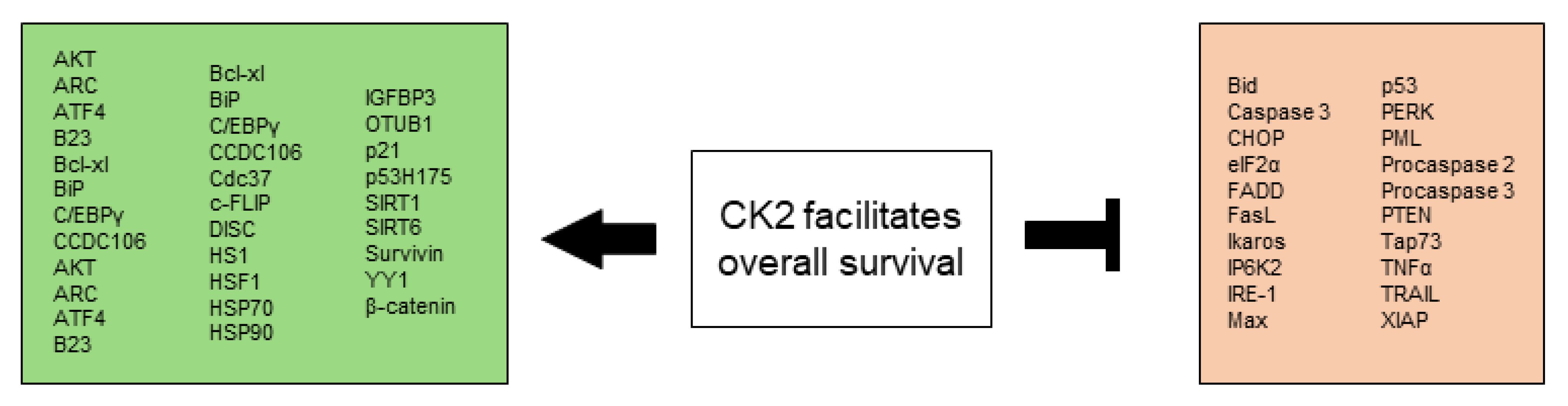
2.2. CK2 Facilitates Altered Stress Response Favoring Overall Survival
2.2.1. CK2 in the Extrinsic Apoptotic Pathway
Influence on Distinct Signal Transducers
Protection from Caspase-Mediated Proteolysis
Influence on Inhibitors of Apoptosis and Growth Factors
2.2.2. CK2 in the Intrinsic Apoptotic Pathway
Influence on Tumor Suppressors and Distinct Signal Transducers
Counteracting p53-Apoptosis Inducing Functions
CK2 Modifies the Cellular Stress Response
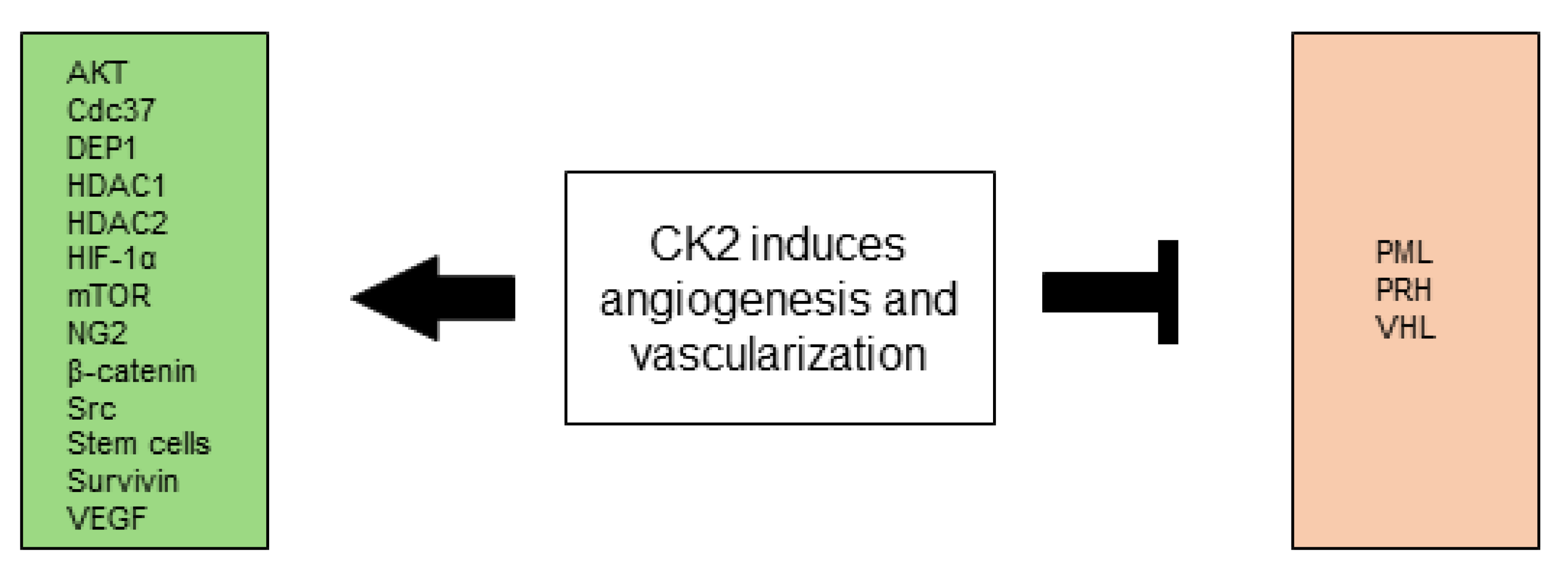
2.3. CK2 Induces Angiogenesis and Vascularization
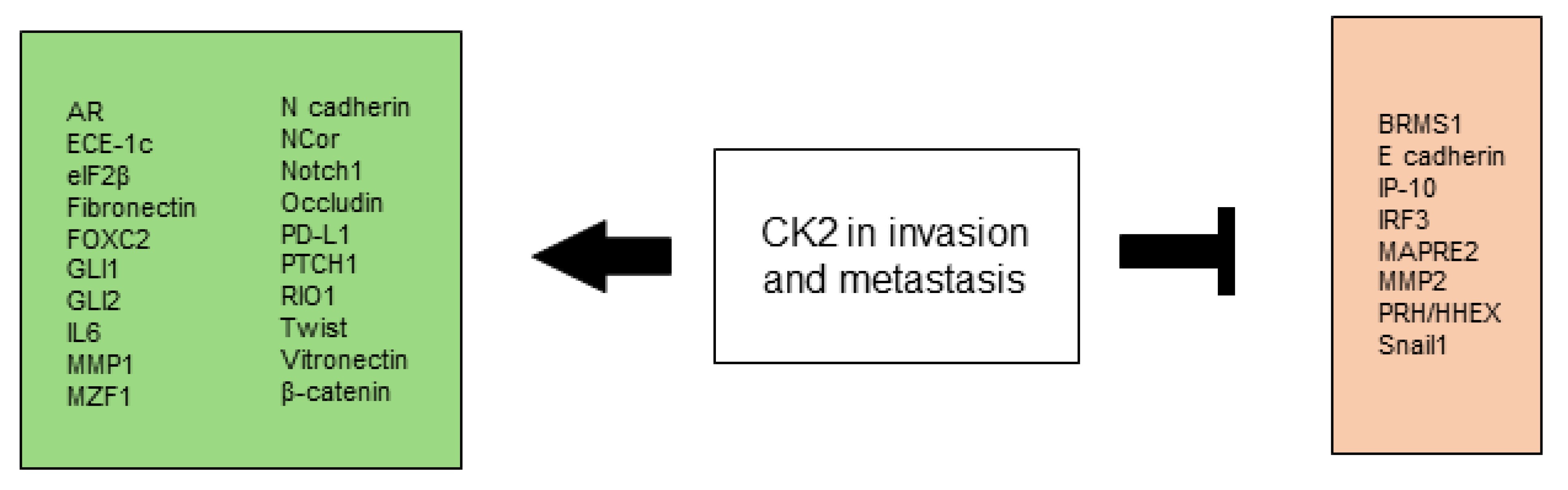
2.4. CK2 Promotes Invasion and Metastasis
2.4.1. Cell Adhesion
2.4.2. Disturbance of mRNA Translation
2.4.3. Disruption of Receptors and Signaling Pathways
2.4.4. Dysregulation of Proteins Normally Relevant in Embryogenesis
2.4.5. Extracellular Matrix
2.4.6. Unbalanced Expression of CK2 Subunits
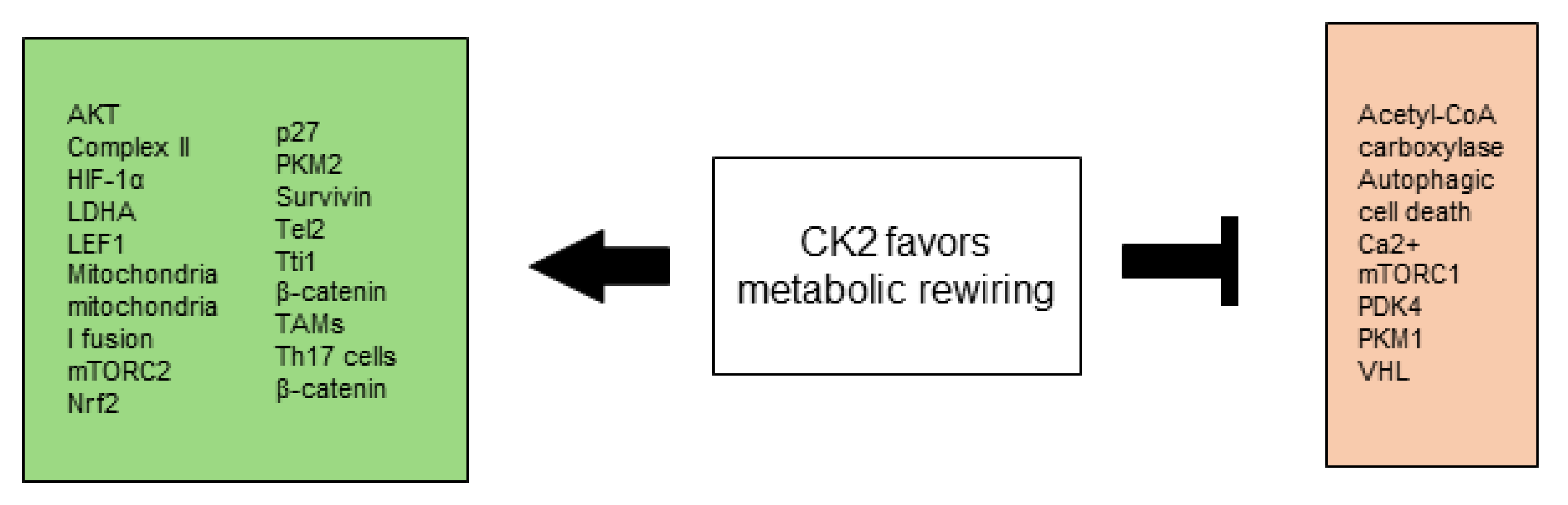
2.5. CK2 Favors Metabolic Rewiring
2.5.1. Warburg Effect
2.5.2. Mitochondrial Metabolism
2.5.3. Autophagy
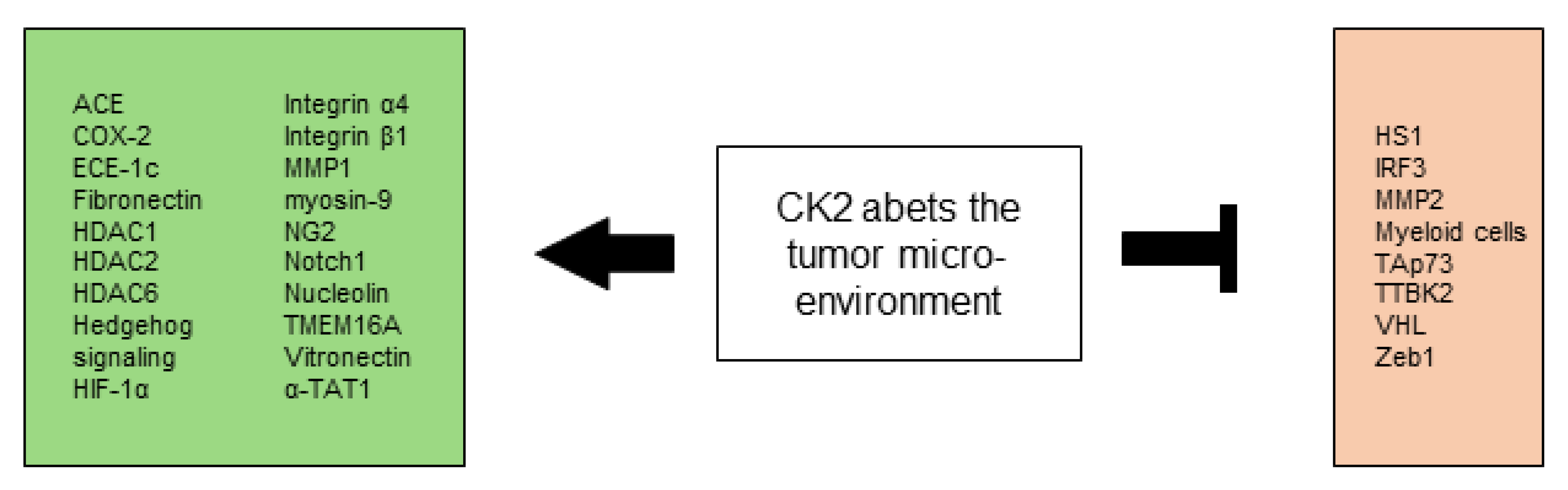
2.6. CK2 Abets the Tumor Microenvironment
2.6.1. Modulation of the Cytoskeleton, Microtubules, and Ion Channels
2.6.2. Influence on Components of the Extracellular Matrix
2.6.3. Cancer Stem Cells
2.6.4. Modulation of the Immune Compartment in the TME
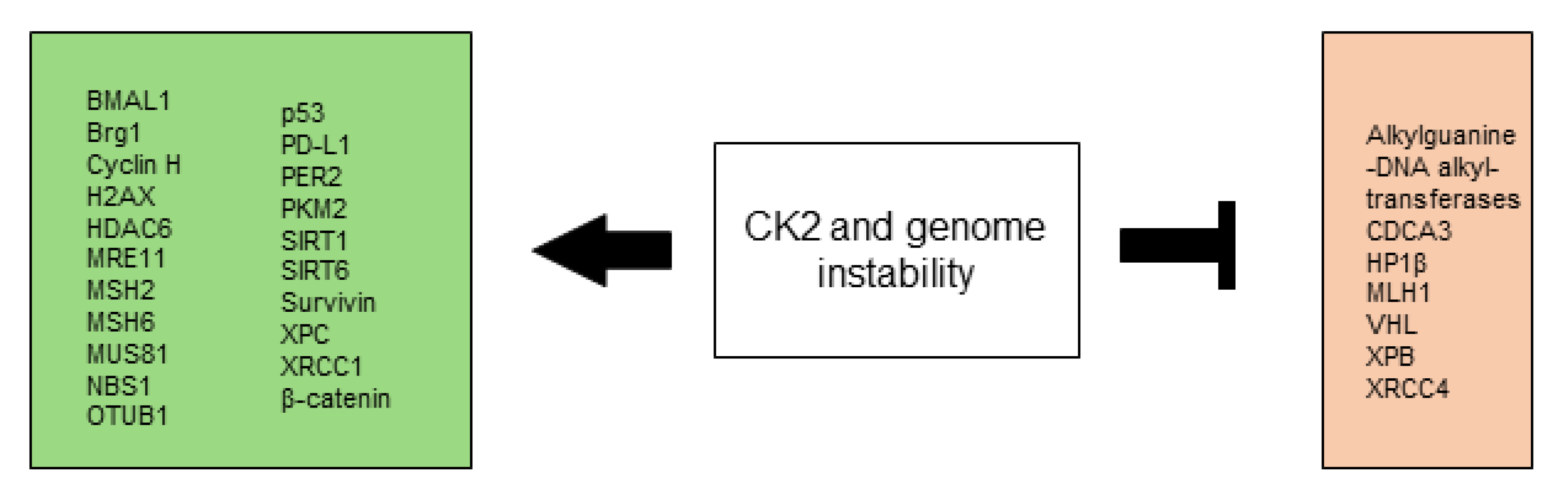
2.7. CK2 in Genome Instability and Mutation
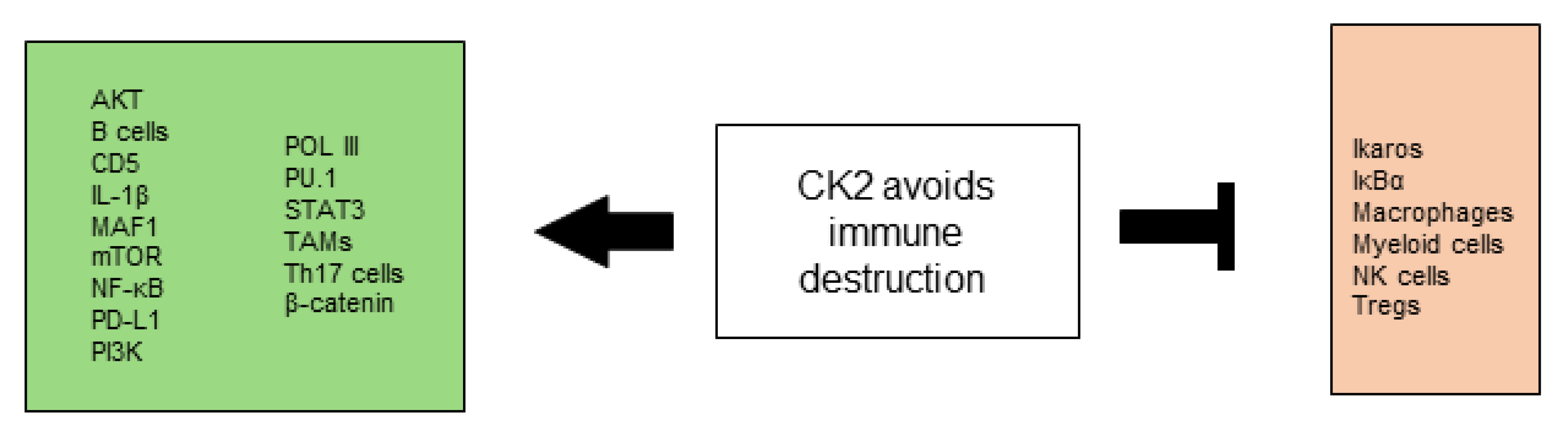
2.8. CK2 Contributes to the Avoidance of Immune Destruction
2.8.1. CK2 and the Innate Immune System
2.8.2. CK2 and the Adaptive Immune System
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Allende, J.E.; Allende, C.C. Protein Kinase CK2: An Enzyme with Multiple Substrates and a Puzzling Regulation. FASEB J. 1995, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- de Villavicencio-Diaz, T.N.; Rabalski, A.J.; Litchfield, D.W. Protein Kinase CK2: Intricate Relationships within Regulatory Cellular Networks. Pharmaceuticals 2017, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W. Protein Kinase CK2: Structure, Regulation and Role in Cellular Decisions of Life and Death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Götz, C.; Montenarh, M. Protein Kinase CK2 in Development and Differentiation. Biomed. Rep. 2017, 6, 127–133. [Google Scholar] [CrossRef]
- Meggio, F.; Pinna, L.A. One-thousand-and-one Substrates of Protein Kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef]
- Gibson, S.A.; Benveniste, E.N. Protein Kinase CK2: An Emerging Regulator of Immunity. Trends Immunol. 2018, 39, 82–85. [Google Scholar] [CrossRef]
- Hong, H.; Benveniste, E.N. The Immune Regulatory Role of Protein Kinase CK2 and Its Implications for Treatment of Cancer. Biomedicines 2021, 9, 1932. [Google Scholar] [CrossRef]
- Montenarh, M. Protein Kinase CK2 in DNA Damage and Repair. Transl. Cancer Res. 2016, 5, 49–63. [Google Scholar] [CrossRef]
- Buchou, T.; Vernet, M.; Blond, O.; Jensen, H.H.; Pointu, H.; Olsen, B.B.; Cochet, C.; Issinger, O.-G.; Boldyreff, B. Disruption of the Regulatory β Subunit of Protein Kinase CK2 in Mice Leads to a Cell-Autonomous Defect and Early Embryonic Lethality. Mol. Cell. Biol. 2003, 23, 908–915. [Google Scholar] [CrossRef]
- Lou, D.Y.; Dominguez, I.; Toselli, P.; Landesman-Bollag, E.; O’Brien, C.; Seldin, D.C. The Alpha Catalytic Subunit of Protein Kinase CK2 Is Required for Mouse Embryonic Development. Mol. Cell. Biol. 2008, 28, 131–139. [Google Scholar] [CrossRef]
- Pinna, L.A. Protein Kinase CK2: A Challenge to Canons. J. Cell Sci. 2002, 115, 3873–3878. [Google Scholar] [CrossRef]
- Chua, M.M.J.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I.; Le Borgne, M. Pharmaceuticals Review CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Roffey, S.E.; Litchfield, D.W. CK2 Regulation: Perspectives in 2021. Biomedicines 2021, 9, 1361. [Google Scholar] [CrossRef]
- Yde, C.W.; Olsen, B.B.; Meek, D.; Watanabe, N.; Guerra, B. The Regulatory β-Subunit of Protein Kinase CK2 Regulates Cell-Cycle Progression at the Onset of Mitosis. Oncogene 2008, 27, 4986–4997. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Nat. Publ. Group 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Bilir, B.; Kucuk, O.; Moreno, C.S. Wnt Signaling Blockage Inhibits Cell Proliferation and Migration, and Induces Apoptosis in Triple-Negative Breast Cancer Cells. J. Transl. Med. 2013, 11, 208. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, H.; Zhu, D. Wnt/β-Catenin Signaling Pathway in Lung Cancer. Med. Drug Discov. 2022, 13, 100113. [Google Scholar] [CrossRef]
- Pei, Y.; Brun, S.N.; Markant, S.L.; Lento, W.; Gibson, P.; Taketo, M.M.; Giovannini, M.; Gilbertson, R.J.; Wechsler-Reya, R.J. WNT Signaling Increases Proliferation and Impairs Differentiation of Stem Cells in the Developing Cerebellum. Development 2012, 139, 1724–1733. [Google Scholar] [CrossRef]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS Production and NF-ΚB Activation Triggered by RAC1 Facilitate WNT-Driven Intestinal Stem Cell Proliferation and Colorectal Cancer Initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68. [Google Scholar] [CrossRef]
- Nusse, R.; Brown, A.; Papkoff, J.; Scambler, P.; Shackleford, G.; McMahon, A.; Moon, R.; Varmus, H. A New Nomenclature for Int-1 and Related Genes: The Wnt Gene Family. Cell 1991, 64, 231. [Google Scholar] [CrossRef]
- Bernatik, O.; Sri Ganji, R.; Dijksterhuis, J.P.; Konik, P.; Cervenka, I.; Polonio, T.; Krejci, P.; Schulte, G.; Bryja, V. Sequential Activation and Inactivation of Dishevelled in the Wnt/Beta-Catenin Pathway by Casein Kinases. J. Biol. Chem. 2011, 286, 10396–10410. [Google Scholar] [CrossRef]
- Seldin, D.C.; Landesman-Bollag, E.; Farago, M.; Currier, N.; Lou, D.; Dominguez, I. CK2 as a Positive Regulator of Wnt Signalling and Tumourigenesis. Mol. Cell. Biochem. 2005, 274, 63–67. [Google Scholar] [CrossRef]
- Song, D.H.; Dominguez, I.; Mizuno, J.; Kaut, M.; Mohr, S.C.; Seldin, D.C. CK2 Phosphorylation of the Armadillo Repeat Region of β-Catenin Potentiates Wnt Signaling. J. Biol. Chem. 2003, 278, 24018–24025. [Google Scholar] [CrossRef]
- Chen, S.; Guttridge, D.C.; You, Z.; Zhang, Z.; Fribley, A.; Mayo, M.W.; Kitajewski, J.; Wang, C.-Y. Wnt-1 Signaling Inhibits Apoptosis by Activating-Catenin/T Cell Factor-Mediated Transcription. J. Cell Biol. 2001, 152, 87–96. [Google Scholar] [CrossRef]
- Song, D.H.; Sussman, D.J.; Seldin, D.C. Endogenous Protein Kinase CK2 Participates in Wnt Signaling in Mammary Epithelial Cells. J. Biol. Chem. 2000, 275, 23790–23797. [Google Scholar] [CrossRef]
- Wang, S.; Jones, K.A. CK2 Controls the Recruitment of Wnt Regulators to Target Genes In Vivo. Curr. Biol. 2006, 16, 2239–2244. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823. [Google Scholar] [CrossRef]
- Cui, X.; Shen, D.; Kong, C.; Zhang, Z.; Zeng, Y.; Lin, X.; Liu, X. NF-ΚB Suppresses Apoptosis and Promotes Bladder Cancer Cell Proliferation by Upregulating Survivin Expression in Vitro and in Vivo. Sci. Rep. 2017, 7, 40723. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-ΚB Activation in Human Breast Cancer Specimens and Its Role in Cell Proliferation and Apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Lyu, Y.L.; Cai, L. NF-ΚB Affects Proliferation and Invasiveness of Breast Cancer Cells by Regulating CD44 Expression. PLoS ONE 2014, 9, e106966. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Qian, H.; Cao, Y.; Zhu, T. Nuclear Factor-ΚB Inhibitor Bay11-7082 Inhibits Gastric Cancer Cell Proliferation by Inhibiting Gli1 Expression. Oncol. Lett. 2021, 21, 301. [Google Scholar] [CrossRef]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced Cell Proliferation by IKK2 Depletion in a Mouse Lung Cancer Model. Nat. Cell Biol. 2012, 14, 257. [Google Scholar] [CrossRef]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional Regulation via the NF-ΚB Signaling Module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef]
- Dominguez, I.; Sonenshein, G.E.; Seldin, D.C. CK2 and Its Role in Wnt and NF-ΚB Signaling: Linking Development and Cancer. Cell. Mol. Life Sci. 2009, 66, 1850–1857. [Google Scholar] [CrossRef]
- Schwarz, E.M.; Van Antwerp, D.; Verma, I.M. Constitutive Phosphorylation of IkappaBalpha by Casein Kinase II Occurs Preferentially at Serine 293: Requirement for Degradation of Free IkappaBalpha. Mol. Cell. Biol. 1996, 16, 3554–3559. [Google Scholar] [CrossRef]
- Kato, T.; Delhase, M.; Hoffmann, A.; Karin, M. CK2 Is a C-Terminal IkappaB Kinase Responsible for NF-KappaB Activation during the UV Response. Mol. Cell 2003, 12, 829–839. [Google Scholar] [CrossRef]
- Biswas, G.; Tang, W.; Sondheimer, N.; Guha, M.; Bansal, S.; Avadhani, N.G. A Distinctive Physiological Role for IκBβ in the Propagation of Mitochondrial Respiratory Stress Signaling. J. Biol. Chem. 2008, 283, 12586. [Google Scholar] [CrossRef]
- Shimada, T.; Kawai, T.; Takeda, K.; Matsumoto, M.; Inoue, J.I.; Tatsumi, Y.; Kanamaru, A.; Akira, S. IKK-i, a Novel Lipopolysaccharide-Inducible Kinase That Is Related to IkappaB Kinases. Int. Immunol. 1999, 11, 1357–1362. [Google Scholar] [CrossRef]
- Eddy, S.F.; Guo, S.; Demicco, E.G.; Romieu-Mourez, R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. Inducible IκB Kinase/IκB Kinase ε Expression Is Induced by CK2 and Promotes Aberrant Nuclear Factor-ΚB Activation in Breast Cancer Cells. Cancer Res. 2005, 65, 11375–11383. [Google Scholar] [CrossRef]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S. Tumor Necrosis Factor α-Induced Phosphorylation of RelA/P65 on Ser529 Is Controlled by Casein Kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef]
- Song, J.; Bae, Y.S. CK2 Down-Regulation Increases the Expression of Senescence-Associated Secretory Phenotype Factors through NF-ΚB Activation. Int. J. Mol. Sci. 2021, 22, 406. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, B.; Liu, Y.; Yu, X.; Cheng, G. Dual Effects of Active ERK in Cancer: A Potential Target for Enhancing Radiosensitivity. Oncol. Lett. 2020, 20, 993. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Ritt, D.A.; Zhou, M.; Conrads, T.P.; Veenstra, T.D.; Copeland, T.D.; Morrison, D.K. CK2 Is a Component of the KSR1 Scaffold Complex That Contributes to Raf Kinase Activation. Curr. Biol. CB 2007, 17, 179–184. [Google Scholar] [CrossRef]
- McKay, M.M.; Morrison, D.K. Integrating Signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef]
- Plotnikov, A.; Chuderland, D.; Karamansha, Y.; Livnah, O.; Seger, R. Nuclear ERK Translocation Is Mediated by Protein Kinase CK2 and Accelerated by Autophosphorylation. Cell. Physiol. Biochem. 2019, 53, 366–387. [Google Scholar] [CrossRef]
- Whitmarsh, A.J. Casein Kinase 2 Sends Extracellular Signal-Regulated Kinase Nuclear. Mol. Cell. Biol. 2011, 31, 3512. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS Signalling Pathways in Cancer Therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Zhou, B.; Ritt, D.A.; Morrison, D.K.; Der, C.J.; Cox, A.D. Protein Kinase CK2α Maintains Extracellular Signal-Regulated Kinase (ERK) Activity in a CK2α Kinase-Independent Manner to Promote Resistance to Inhibitors of RAF and MEK but Not ERK in BRAF Mutant Melanoma. J. Biol. Chem. 2016, 291, 17804–17815. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT Signaling Pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and Consequences of Jak–STAT Signaling in the Immune System. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The Role of JAK-STAT Signaling Pathway and Its Regulators in the Fate of T Helper Cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein Kinase CK2: A Potential Therapeutic Target for Diverse Human Diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef]
- Zheng, Y.; McFarland, B.C.; Drygin, D.; Yu, H.; Bellis, S.L.; Kim, H.; Bredel, M.; Benveniste, E.N. Targeting Protein Kinase CK2 Suppresses Prosurvival Signaling Pathways and Growth of Glioblastoma. Clin. Cancer Res. 2013, 19, 6484–6494. [Google Scholar] [CrossRef]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.T.; Li, J.; Sha, B.; et al. ACK2-Dependent Mechanism for Activation of the JAK-STAT Signaling Pathway. Blood 2011, 118, 156–166. [Google Scholar] [CrossRef]
- Manni, S.; Brancalion, A.; Mandato, E.; Tubi, L.Q.; Colpo, A.; Pizzi, M.; Cappellesso, R.; Zaffino, F.; Antonia, S.; Maggio, D.; et al. Protein Kinase CK2 Inhibition Down Modulates the NF-KB and STAT3 Survival Pathways, Enhances the Cellular Proteotoxic Stress and Synergistically Boosts the Cytotoxic Effect of Bortezomib on Multiple Myeloma and Mantle Cell Lymphoma Cells. PLoS ONE 2013, 8, e75280. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of Interleukin-6 in Cancer Progression and Therapeutic Resistance. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Brooks, A.J.; Putoczki, T. JAK-STAT Signalling Pathway in Cancer. Cancers 2020, 12, 1971. [Google Scholar] [CrossRef] [PubMed]
- Drygin, D.; Ho, C.B.; Omori, M.; Bliesath, J.; Proffitt, C.; Rice, R.; Siddiqui-Jain, A.; O’Brien, S.; Padgett, C.; Lim, J.K.C.; et al. Protein Kinase CK2 Modulates IL-6 Expression in Inflammatory Breast Cancer. Biochem. Biophys. Res. Commun. 2011, 415, 163–167. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The Role of STAT3 in Autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Mandal, T.; Bhowmik, A.; Chatterjee, A.; Chatterjee, U.; Chatterjee, S.; Ghosh, M.K. Reduced Phosphorylation of Stat3 at Ser-727 Mediated by Casein Kinase 2—Protein Phosphatase 2A Enhances Stat3 Tyr-705 Induced Tumorigenic Potential of Glioma Cells. Cell. Signal. 2014, 26, 1725–1734. [Google Scholar] [CrossRef]
- Hériché, J.K.; Lebrin, F.; Rabilloud, T.; Leroy, D.; Chambaz, E.M.; Goldberg, Y. Regulation of Protein Phosphatase 2A by Direct Interaction with Casein Kinase 2alpha. Science 1997, 276, 952–955. [Google Scholar] [CrossRef]
- Nho, R.S.; Hergert, P. FoxO3a and Disease Progression. World J. Biol. Chem. 2014, 5, 346. [Google Scholar] [CrossRef]
- Qian, C.; Liu, Q. FOXO3a Inhibits Nephroblastoma Cell Proliferation, Migration and Invasion, and Induces Apoptosis through Downregulating the Wnt/Β-catenin Signaling Pathway. Mol. Med. Rep. 2021, 24, 796. [Google Scholar] [CrossRef]
- Korashy, H.M.; Belali, O.M.; Ansar, M.A.; Alharbi, N.O. FoxO3a Is Essential for the Antiproliferative and Apoptogenic Effects of Sunitinib in MDA-MB231 Cell Line. Anticancer. Res. 2016, 36, 6097–6108. [Google Scholar] [CrossRef]
- Poulsen, R.C.; Carr, A.J.; Hulley, P.A. Cell Proliferation Is a Key Determinant of the Outcome of FOXO3a Activation. Biochem. Biophys. Res. Commun. 2015, 462, 78–84. [Google Scholar] [CrossRef]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical Role of FOXO3a in Carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef]
- Torres, J.; Pulido, R. The Tumor Suppressor PTEN Is Phosphorylated by the Protein Kinase CK2 at Its C Terminus Implications for Pten Stability to Proteasome-Mediated Degradation. J. Biol. Chem. 2000, 276, 993–998. [Google Scholar] [CrossRef]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN Tail Regulates Protein Stability and Function. Mol. Cell. Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein Kinase CK2 Phosphorylates and Upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef]
- Nakamura, N.; Ramaswamy, S.; Vazquez, F.; Signoretti, S.; Loda, M.; Sellers, W.R. Forkhead Transcription Factors Are Critical Effectors of Cell Death and Cell Cycle Arrest Downstream of PTEN. Mol. Cell. Biol. 2000, 20, 8969. [Google Scholar] [CrossRef]
- Chatterjee, A.; Chatterjee, U.; Ghosh, M.K. Activation of Protein Kinase CK2 Attenuates FOXO3a Functioning in a PML-Dependent Manner: Implications in Human Prostate Cancer. Cell Death Dis. 2013, 4, e543. [Google Scholar] [CrossRef]
- Soufi, A.; Noy, P.; Buckle, M.; Sawasdichai, A.; Gaston, K.; Jayaraman, P.S. CK2 Phosphorylation of the PRH/Hex Homeodomain Functions as a Reversible Switch for DNA Binding. Nucleic Acids Res. 2009, 37, 3288–3300. [Google Scholar] [CrossRef]
- Siddiqui, Y.H.; Kershaw, R.M.; Humphreys, E.H.; Assis Junior, E.M.; Chaudhri, S.; Jayaraman, P.S.; Gaston, K. CK2 Abrogates the Inhibitory Effects of PRH/HHEX on Prostate Cancer Cell Migration and Invasion and Acts through PRH to Control Cell Proliferation. Oncogenesis 2017, 6, e293. [Google Scholar] [CrossRef]
- Noy, P.; Sawasdichai, A.; Jayaraman, P.S.; Gaston, K. Protein Kinase CK2 Inactivates PRH/Hhex Using Multiple Mechanisms to de-Repress VEGF-Signalling Genes and Promote Cell Survival. Nucleic Acids Res. 2012, 40, 9008–9020. [Google Scholar] [CrossRef]
- Catena, V.; Bruno, T.; Iezzi, S.; Matteoni, S.; Salis, A.; Sorino, C.; Damonte, G.; Fanciulli, M. CK2-Mediated Phosphorylation of Che-1/AATF Is Required for Its pro-Proliferative Activity. J. Exp. Clin. Cancer Res. 2021, 40, 232. [Google Scholar] [CrossRef]
- Ali, M.U.; Ur Rahman, M.S.; Jia, Z.; Jiang, C. Eukaryotic Translation Initiation Factors and Cancer. Tumor Biol. 2017, 39, 1010428317709805. [Google Scholar] [CrossRef]
- Homma, M.K.; Wada, I.; Suzuki, T.; Yamaki, J.; Krebs, E.G.; Homma, Y. CK2 Phosphorylation of Eukaryotic Translation Initiation Factor 5 Potentiates Cell Cycle Progression. Proc. Natl. Acad. Sci. USA 2005, 102, 15688–15693. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Bliesath, J.; Huser, N.; Omori, M.; Bunag, D.; Proffitt, C.; Streiner, N.; Ho, C.; Siddiqui-Jain, A.; O’Brien, S.E.; Lim, J.K.C.; et al. Combined Inhibition of EGFR and CK2 Augments the Attenuation of PI3K-Akt-MTOR Signaling and the Killing of Cancer Cells. Cancer Lett. 2012, 322, 113–118. [Google Scholar] [CrossRef]
- Chou, S.T.; Patil, R.; Galstyan, A.; Gangalum, P.R.; Cavenee, W.K.; Furnari, F.B.; Ljubimov, V.A.; Chesnokova, A.; Kramerov, A.A.; Ding, H.; et al. Simultaneous Blockade of Interacting CK2 and EGFR Pathways by Tumor-Targeting Nanobioconjugates Increases Therapeutic Efficacy against Glioblastoma Multiforme. J. Control. Release 2016, 244, 14–23. [Google Scholar] [CrossRef] [PubMed]
- So, K.S.; Kim, C.H.; Rho, J.K.; Kim, S.Y.; Choi, Y.J.; Song, J.S.; Kim, W.S.; Choi, C.M.; Chun, Y.J.; Lee, J.C. Autophagosome-Mediated EGFR Down-Regulation Induced by the CK2 Inhibitor Enhances the Efficacy of EGFR-TKI on EGFR-Mutant Lung Cancer Cells with Resistance by T790M. PLoS ONE 2014, 9, e114000. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhou, F.; Zhou, Y.; Zhang, S.; Li, Q.; Li, Z.; Liu, L.; Wu, G.; Meng, R. Quinalizarin, a Specific CK2 Inhibitor, Can Reduce Icotinib Resistance in Human Lung Adenocarcinoma Cell Lines. Int. J. Mol. Med. 2019, 44, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Sherman, M.E.; Kannan, N.; Stanczyk, F.Z. Progesterone and Breast Cancer. Endocr. Rev. 2020, 41, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Lange, C.A. Progesterone Receptors Upregulate Wnt-1 to Induce Epidermal Growth Factor Receptor Transactivation and c-Src-Dependent Sustained Activation of Erk1/2 Mitogen-Activated Protein Kinase in Breast Cancer Cells. Mol. Cell. Biol. 2007, 27, 466–480. [Google Scholar] [CrossRef]
- Zhang, Y.; Beck, C.A.; Poletti, A.; Edwards, D.P.; Weigel, N.L. Identification of Phosphorylation Sites Unique to the B Form of Human Progesterone Receptor. In Vitro Phosphorylation by Casein Kinase II. J. Biol. Chem. 1994, 269, 31034–31040. [Google Scholar] [CrossRef]
- Hagan, C.R.; Regan, T.M.; Dressing, G.E.; Lange, C.A. Ck2-Dependent Phosphorylation of Progesterone Receptors (PR) on Ser81 Regulates PR-B Isoform-Specific Target Gene Expression in Breast Cancer Cells. Mol. Cell. Biol. 2011, 31, 2439–2452. [Google Scholar] [CrossRef]
- Cobb, L.J.; Mehta, H.; Cohen, P. Enhancing the Apoptotic Potential of Insulin-Like Growth Factor-Binding Protein-3 in Prostate Cancer by Modulation of CK2 Phosphorylation. Mol. Endocrinol. 2009, 23, 1624–1633. [Google Scholar] [CrossRef]
- Varma Shrivastav, S.; Bhardwaj, A.; Pathak, K.A.; Shrivastav, A. Insulin-Like Growth Factor Binding Protein-3 (IGFBP-3): Unraveling the Role in Mediating IGF-Independent Effects Within the Cell. Front. Cell Dev. Biol. 2020, 8, 286. [Google Scholar] [CrossRef]
- Kumar, R.; Gururaj, A.E.; Barnes, C.J. P21-Activated Kinases in Cancer. Nat. Rev. Cancer 2006, 6, 459–471. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kim, Y.B.; Kim, J.H. Protein Kinase CK2 Phosphorylates and Activates P21-Activated Kinase 1. Mol. Biol. Cell 2013, 24, 2990–2999. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 Chaperone Machinery. Nat. Reviews. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Xu, Q.; Tu, J.; Dou, C.; Zhang, J.; Yang, L.; Liu, X.; Lei, K.; Liu, Z.; Wang, Y.; Li, L.; et al. HSP90 Promotes Cell Glycolysis, Proliferation and Inhibits Apoptosis by Regulating PKM2 Abundance via Thr-328 Phosphorylation in Hepatocellular Carcinoma. Mol. Cancer 2017, 16, 178. [Google Scholar] [CrossRef]
- Vartholomaiou, E.; Echeverría, P.C.; Picard, D. Unusual Suspects in the Twilight Zone Between the Hsp90 Interactome and Carcinogenesis. Adv. Cancer Res. 2016, 129, 1–30. [Google Scholar] [CrossRef]
- Longshaw, V.M.; Chapple, J.P.; Balda, M.S.; Cheetham, M.E.; Blatch, G.L. Nuclear Translocation of the Hsp70/Hsp90 Organizing Protein MSTI1 Is Regulated by Cell Cycle Kinases. J. Cell Sci. 2004, 117, 701–710. [Google Scholar] [CrossRef]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef]
- Muller, P.; Ruckova, E.; Halada, P.; Coates, P.J.; Hrstka, R.; Lane, D.P.; Vojtesek, B. C-Terminal Phosphorylation of Hsp70 and Hsp90 Regulates Alternate Binding to Co-Chaperones CHIP and HOP to Determine Cellular Protein Folding/Degradation Balances. Oncogene 2012, 32, 3101–3110. [Google Scholar] [CrossRef]
- Tsai, C.L.; Chao, A.S.; Jung, S.M.; Lin, C.Y.; Chao, A.; Wang, T.H. Stress-Induced Phosphoprotein 1 Acts as a Scaffold Protein for Glycogen Synthase Kinase-3 Beta-Mediated Phosphorylation of Lysine-Specific Demethylase 1. Oncogenesis 2018, 7, 1–17. [Google Scholar] [CrossRef]
- Lv, T.; Yuan, D.; Miao, X.; Lv, Y.; Zhan, P.; Shen, X.; Song, Y. Over-Expression of LSD1 Promotes Proliferation, Migration and Invasion in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e35065. [Google Scholar] [CrossRef]
- Miyata, Y. Protein Kinase CK2 in Health and Disease. Cell. Mol. Life Sci. 2009, 66, 1840–1849. [Google Scholar] [CrossRef]
- Sang Bae, J.; Park, S.-H.; Min Kim, K.; Sang Kwon, K.; Young Kim, C.; Ku Lee, H.; Park, B.-H.; Sung Park, H.; Lee, H.; Sung Moon, W.; et al. CK2a Phosphorylates DBC1 and Is Involved in the Progression of Gastric Carcinoma and Predicts Poor Survival of Gastric Carcinoma Patients. UICC Int. J. Cancer IJC 2015, 136, 797–809. [Google Scholar] [CrossRef]
- Fang, Q.; Bellanti, J.A.; Zheng, S.G. Advances on the Role of the Deleted in Breast Cancer (DBC1) in Cancer and Autoimmune Diseases. J. Leukoc. Biol. 2021, 109, 449–454. [Google Scholar] [CrossRef]
- Zschoernig, B.; Mahlknecht, U. Carboxy-Terminal Phosphorylation of SIRT1 by Protein Kinase CK2. Biochem. Biophys. Res. Commun. 2009, 381, 372–377. [Google Scholar] [CrossRef]
- Krężel, A.; Maret, W. The Biological Inorganic Chemistry of Zinc Ions. Arch. Biochem. Biophys. 2016, 611, 3–19. [Google Scholar] [CrossRef]
- Myers, S.A.; Nield, A.; Chew, G.S.; Myers, M.A. The Zinc Transporter, Slc39a7 (Zip7) Is Implicated in Glycaemic Control in Skeletal Muscle Cells. PLoS ONE 2013, 8, e79316. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, H.; Xu, Z.; Cheng, X. Zinc Dysregulation in Cancers and Its Potential as a Therapeutic Target. Cancer Biol. Med. 2020, 17, 612. [Google Scholar] [CrossRef]
- Taylor, K.M.; Hiscox, S.; Nicholson, R.I.; Hogstrand, C.; Kille, P. Cell Biology: Protein Kinase CK2 Triggers Cytosolic Zinc Signaling Pathways by Phosphorylation of Zinc Channel ZIP7. Sci. Signal. 2012, 5, 8–10. [Google Scholar] [CrossRef]
- Bafaro, E.; Liu, Y.; Xu, Y.; Dempski, R.E. The Emerging Role of Zinc Transporters in Cellular Homeostasis and Cancer. Signal Transduct. Target. Ther. 2017, 2, 17029. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, A.; Dubé, N.; Tremblay, M.L. Cytoplasmic Protein Tyrosine Phosphatases, Regulation and Function: The Roles of PTP1B and TC-PTP. Curr. Opin. Cell Biol. 2005, 17, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Sheng, N.; Yan, L.; You, W.; Tan, G.; Gong, J.; Chen, H.; Yang, Y.; Hu, L.; Wang, Z. Knockdown of SLC39A7 Inhibits Cell Growth and Induces Apoptosis in Human Colorectal Cancer Cells. Acta Biochim. Et Biophys. Sin. 2017, 49, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Vichova, P.; Jordan, N.; Hiscox, S.; Hendley, R.; Nicholson, R.I. ZIP7-Mediated Intracellular Zinc Transport Contributes to Aberrant Growth Factor Signaling in Antihormone-Resistant Breast Cancer Cells. Endocrinology 2008, 149, 4912–4920. [Google Scholar] [CrossRef]
- Zaman, M.S.; Johnson, A.J.; Petersingham, G.; Muench, G.W.; Dong, Q.; Wu, M.J. Protein Kinase CK2 Is Involved in Zinc Homeostasis in Breast and Prostate Cancer Cells. BioMetals 2019, 32, 861–873. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 Apoptotic Switch in Cancer Development and Therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495. [Google Scholar] [CrossRef]
- Wang, G.; Ahmad, K.A.; Ahmed, K. Modulation of Death Receptor-Mediated Apoptosis by CK2. Mol. Cell. Biochem. 2005, 274, 201–205. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, B. Mechanisms of Resistance to TRAIL-Induced Apoptosis in Cancer. Cancer Gene Ther. 2004, 12, 228–237. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Rue, M.; Comella, J.X.; Matias-Guiu, X. FLIP Is Frequently Expressed in Endometrial Carcinoma and Has a Role in Resistance to TRAIL-Induced Apoptosis. Lab. Investig. A J. Tech. Methods Pathol. 2005, 85, 885–894. [Google Scholar] [CrossRef]
- Llobet, D.; Eritja, N.; Encinas, M.; Llecha, N.; Yeramian, A.; Pallares, J.; Sorolla, A.; Gonzalez-Tallada, F.J.; Matias-Guiu, X.; Dolcet, X. CK2 Controls TRAIL and Fas Sensitivity by Regulating FLIP Levels in Endometrial Carcinoma Cells. Oncogene 2007, 27, 2513–2524. [Google Scholar] [CrossRef]
- Vilmont, V.; Filhol, O.; Hesse, A.M.; Couté, Y.; Hue, C.; Rémy-Tourneur, L.; Mistou, S.; Cochet, C.; Chiocchia, G. Modulatory Role of the Anti-Apoptotic Protein Kinase CK2 in the Sub-Cellular Localization of Fas Associated Death Domain Protein (FADD). Biochim. Et Biophys. Acta-Mol. Cell Res. 2015, 1853, 2885–2896. [Google Scholar] [CrossRef]
- Izeradjene, K.; Douglas, L.; Delaney, A.; Houghton, J.A. Influence of Casein Kinase II in Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Induced Apoptosis in Human Rhabdomyosarcoma Cells. Clin. Cancer Res. 2004, 10, 6650–6660. [Google Scholar] [CrossRef]
- Filhol, O.; Cochet, C. Protein Kinases Curb Cell Death. Sci. Signal. 2011, 4, pe26. [Google Scholar] [CrossRef]
- Duncan, J.S.; Turowec, J.P.; Vilk, G.; Li, S.S.C.; Gloor, G.B.; Litchfield, D.W. Regulation of Cell Proliferation and Survival: Convergence of Protein Kinases and Caspases. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2010, 1804, 505–510. [Google Scholar] [CrossRef]
- Duncan, J.S.; Turowec, J.P.; Duncan, K.E.; Vilk, G.; Wu, C.; Lüscher, B.; Li, S.S.C.; Gloor, G.B.; Litchfield, D.W. A Peptide-Based Target Screen Implicates the Protein Kinase CK2 in the Global Regulation of Caspase Signaling. Sci. Signal. 2011, 4, ra30. [Google Scholar] [CrossRef]
- Turowec, J.P.; Vilk, G.; Gabriel, M.; Litchfield, D.W. Characterizing the Convergence of Protein Kinase CK2 and Caspase-3 Reveals Isoform-Specific Phosphorylation of Caspase-3 by CK2a’: Implications for Pathological Roles of CK2 in Promoting Cancer Cell Survival. Oncotarget 2013, 4, 560–571. [Google Scholar] [CrossRef]
- Vigneswara, V.; Ahmed, Z. The Role of Caspase-2 in Regulating Cell Fate. Cells 2020, 9, 1259. [Google Scholar] [CrossRef]
- Terry, M.R.; Arya, R.; Mukhopadhyay, A.; Berrett, K.C.; Clair, P.M.; Witt, B.; Salama, M.E.; Bhutkar, A.; Oliver, T.G. Caspase-2 Impacts Lung Tumorigenesis and Chemotherapy Response in Vivo. Cell Death Differ. 2015, 22, 719–730. [Google Scholar] [CrossRef]
- Puccini, J.; Dorstyn, L.; Kumar, S. Caspase-2 as a Tumour Suppressor. Cell Death Differ. 2013, 20, 1133–1139. [Google Scholar] [CrossRef]
- Shin, S.; Lee, Y.; Kim, W.; Ko, H.; Choi, H.; Kim, K. Caspase-2 Primes Cancer Cells for TRAIL-Mediated Apoptosis by Processing Procaspase-8. EMBO J. 2005, 24, 3532–3542. [Google Scholar] [CrossRef]
- Ruzzene, M.; Brunati, A.M.; Sarno, S.; Marin, O.; Donella-Deana, A.; Pinna, L.A. Ser/Thr Phosphorylation of Hematopoietic Specific Protein 1 (HS1) Implication of Protein Kinase CK2. Eur. J. Biochem. 2000, 267, 3065–3072. [Google Scholar] [CrossRef]
- Ruzzene, M.; Penzo, D.; Pinna, L.A. Protein Kinase CK2 Inhibitor 4,5,6,7-Tetrabromobenzotriazole (TBB) Induces Apoptosis and Caspase-Dependent Degradation of Haematopoietic Lineage Cell-Specific Protein 1 (HS1) in Jurkat Cells. Biochem. J 2002, 364, 41–47. [Google Scholar] [CrossRef]
- Rottner, K.; Stradal, T.E.B. Actin Dynamics and Turnover in Cell Motility. Curr. Opin. Cell Biol. 2011, 23, 569–578. [Google Scholar] [CrossRef]
- Collins, M.K.L.; Perkins, G.R.; Rodriguez-Tarduchy, G.; Nieto, M.A.; López-Rivas, A. Growth Factors as Survival Factors: Regulation of Apoptosis. Bioessays 1994, 16, 133–138. [Google Scholar] [CrossRef]
- Lacasse, E.C.; Baird, S.; Korneluk, R.G.; Mackenzie, A.E. The Inhibitors of Apoptosis (IAPs) and Their Emerging Role in Cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar] [CrossRef]
- Ludwig-Galezowska, A.H.; Flanagan, L.; Rehm, M. Apoptosis Repressor with Caspase Recruitment Domain, a Multifunctional Modulator of Cell Death. J. Cell. Mol. Med. 2011, 15, 1044. [Google Scholar] [CrossRef]
- Li, P.F.; Li, J.; Müller, E.C.; Otto, A.; Dietz, R.; Von Harsdorf, R. Phosphorylation by Protein Kinase CK2: A Signaling Switch for the Caspase-Inhibiting Protein ARC. Mol. Cell 2002, 10, 247–258. [Google Scholar] [CrossRef]
- Wang, J.; Feng, C.; He, Y.; Ding, W.; Sheng, J.; Arshad, M.; Zhang, X.; Li, P. Phosphorylation of Apoptosis Repressor with Caspase Recruitment Domain by Protein Kinase CK2 Contributes to Chemotherapy Resistance by Inhibiting Doxorubicin Induced Apoptosis. Oncotarget 2015, 6, 27700. [Google Scholar] [CrossRef]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A Molecular Biomarker in Cancer. Indian J. Med. Res. 2015, 141, 389. [Google Scholar] [CrossRef]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A Unique Target for Tumor Therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- He, S.Q.; Rehman, H.; Gong, M.G.; Zhao, Y.Z.; Huang, Z.Y.; Li, C.H.; Zhang, W.G.; Chen, X.P. Inhibiting Survivin Expression Enhances TRAIL-Induced Tumoricidal Activity in Human Hepatocellular Carcinoma via Cell Cycle Arrest. Cancer Biol. Ther. 2007, 6, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Azuhata, T.; Scott, D.; Griffith, T.S.; Miller, M.; Sandler, A.D. Survivin Inhibits Apoptosis Induced by TRAIL, and the Ratio between Survivin and TRAIL Receptors Is Predictive of Recurrent Disease in Neuroblastoma. J. Pediatric Surg. 2006, 41, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.C.; Torres, V.A.; Rodriguez, D.A.; Leyton, L.; Quest, A.F.G. Casein Kinase 2 (CK2) Increases Survivin Expression via Enhanced Beta-Catenin-T Cell Factor/Lymphoid Enhancer Binding Factor-Dependent Transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 15079–15084. [Google Scholar] [CrossRef]
- Fernández, J.G.; Rodríguez, D.A.; Valenzuela, M.; Calderon, C.; Urzúa, U.; Munroe, D.; Rosas, C.; Lemus, D.; Díaz, N.; Wright, M.C.; et al. Survivin Expression Promotes VEGF-Induced Tumor Angiogenesis via PI3K/Akt Enhanced β-Catenin/Tcf-Lef Dependent Transcription. Mol. Cancer 2014, 13, 1–15. [Google Scholar] [CrossRef]
- Barrett, R.M.A.; Colnaghi, R.; Wheatley, S.P. Threonine 48 in the BIR Domain of Survivin Is Critical to Its Mitotic and Anti-Apoptotic Activities and Can Be Phosphorylated by CK2 in Vitro. Cell Cycle 2011, 10, 538–548. [Google Scholar] [CrossRef]
- Gassmann, R.; Carvalho, A.; Henzing, A.J.; Ruchaud, S.; Hudson, D.F.; Honda, R.; Nigg, E.A.; Gerloff, D.L.; Earnshaw, W.C. Borealin: A Novel Chromosomal Passenger Required for Stability of the Bipolar Mitotic Spindle. J. Cell Biol. 2004, 166, 179. [Google Scholar] [CrossRef]
- Barata, J.T. The Impact of PTEN Regulation by CK2 on PI3K-Dependent Signaling and Leukemia Cell Survival. Adv. Enzym. Regul. 2011, 51, 37–49. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Incani, U.C.; Curatolo, A.D.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef]
- Weng, L.P.; Brown, J.L.; Eng, C. PTEN Induces Apoptosis and Cell Cycle Arrest through Phosphoinositol-3-Kinase/Akt-Dependent and -Independent Pathways. Hum. Mol. Genet. 2001, 10, 237–242. [Google Scholar] [CrossRef]
- Miller, S.J.; Lou, D.Y.; Seldin, D.C.; Lane, W.S.; Neel, B.G. Direct Identification of PTEN Phosphorylation Sites. FEBS Lett. 2002, 528, 145–153. [Google Scholar] [CrossRef]
- Torres, J.; Rodriguez, J.; Myers, M.P.; Valiente, M.; Graves, J.D.; Tonks, N.K.; Pulido, R. Phosphorylation-Regulated Cleavage of the Tumor Suppressor PTEN by Caspase-3 Implications for the Control of Protein Stability and Pten-Protein Interactions. J. Biol. Chem. 2003, 278, 30652–30660. [Google Scholar] [CrossRef]
- Ross, A.H.; Gericke, A. Phosphorylation Keeps PTEN Phosphatase Closed for Business. Proc. Natl. Acad. Sci. USA 2009, 106, 1297–1298. [Google Scholar] [CrossRef]
- Odriozola, L.; Singh, G.; Hoang, T.; Chan, A.M. Regulation of PTEN Activity by Its Carboxyl-Terminal Autoinhibitory Domain. J. Biol. Chem. 2007, 282, 23306–23315. [Google Scholar] [CrossRef]
- Hsu, K.S.; Kao, H.Y. PML: Regulation and Multifaceted Function beyond Tumor Suppression. Cell Biosci. 2018, 8, 5. [Google Scholar] [CrossRef]
- Guan, D.; Kao, H.-Y. The Function, Regulation and Therapeutic Implications of the Tumor Suppressor Protein, PML. Cell Biosci. 2015, 5, 60. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Pandolfi, P.P. The Role of PML in the Control of Apoptotic Cell Fate: A New Key Player at ER-Mitochondria Sites. Cell Death Differ. 2011, 18, 1450–1456. [Google Scholar] [CrossRef]
- Scaglioni, P.P.; Yung, T.M.; Cai, L.F.; Erdjument-Bromage, H.; Kaufman, A.J.; Singh, B.; Teruya-Feldstein, J.; Tempst, P.; Pandolfi, P.P. A CK2-Dependent Mechanism for Degradation of the PML Tumor Suppressor. Cell 2006, 126, 269–283. [Google Scholar] [CrossRef]
- Götz, C.; Wagner, P.; Issinger, O.G.; Montenarh, M. P21WAF1/CIP1 Interacts with Protein Kinase CK2. Oncogene 1996, 13, 391–398. [Google Scholar]
- Götz, C.; Kartarius, S.; Scholtes, P.; Montenarh, M. Binding Domain for P21(WAF1) on the Polypeptide Chain of the Protein Kinase CK2 Beta-Subunit. Biochem. Biophys. Res. Commun. 2000, 268, 882–885. [Google Scholar] [CrossRef]
- Romero-Oliva, F.; Allende, J.E. Protein P21 WAF1/CIP1 Is Phosphorylated by Protein Kinase CK2 In Vitro and Interacts with the Amino Terminal End of the CK2 Beta Subunit. J. Cell. Biochem. 2001, 81, 445–452. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple Functions of P21 in Cell Cycle, Apoptosis and Transcriptional Regulation after DNA Damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate Networks and Multiple Activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Zhou, B.P.; Liao, Y.; Xia, W.; Spohn, B.; Lee, M.H.; Hung, M.C. Cytoplasmic Localization of P21Cip1/WAF1 by Akt-Induced Phosphorylation in HER-2/Neu-Overexpressing Cells. Nat. Cell Biol. 2001, 3, 245–252. [Google Scholar] [CrossRef]
- Li, Y.; Dowbenko, D.; Lasky, L.A. AKT/PKB Phosphorylation of P21Cip/WAF1 Enhances Protein Stability of P21Cip/WAF1 and Promotes Cell Survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef] [PubMed]
- Bui, N.L.C.; Pandey, V.; Zhu, T.; Ma, L.; Basappa; Lobie, P.E. Bad Phosphorylation as a Target of Inhibition in Oncology. Cancer Lett. 2018, 415, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, S.; Mäurer, A.; Zhu, Y.; Aichele, D.; Pinna, L.A.; Krieglstein, J. Protein Kinase CK2 Phosphorylates BAD at Threonine-117. Neurochem. Int. 2004, 45, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Green, M.M.L.; Hutchison, G.J.; Valentine, H.R.; Fitzmaurice, R.J.; Davidson, S.E.; Hunter, R.D.; Dive, C.; West, C.M.L.; Stratford, I.J. Expression of the Proapoptotic Protein Bid Is an Adverse Prognostic Factor for Radiotherapy Outcome in Carcinoma of the Cervix. Br. J. Cancer 2005, 92, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, M.; Ferry, G.; Masdehors, P.; Boutin, J.A.; Hickman, J.A.; Dive, C. Post-Translational Modification of Bid Has Differential Effects on Its Susceptibility to Cleavage by Caspase 8 or Caspase 3. J. Biol. Chem. 2003, 278, 15749–15757. [Google Scholar] [CrossRef]
- Desagher, S.; Osen-Sand, A.; Montessuit, S.; Magnenat, E.; Vilbois, F.; Hochmann, A.; Journot, L.; Antonsson, B.; Martinou, J.C. Phosphorylation of Bid by Casein Kinases I and II Regulates Its Cleavage by Caspase 8. Mol. Cell 2001, 8, 601–611. [Google Scholar] [CrossRef]
- Olsen, B.B.; Petersen, J.; Issinger, O.G. BID, an Interaction Partner of Protein Kinase CK2α. Biol. Chem. 2006, 387, 441–449. [Google Scholar] [CrossRef]
- Hellwig, C.T.; Ludwig-Galezowska, A.H.; Concannon, C.G.; Litchfield, D.W.; Prehn, J.H.M.; Rehm, M. Activity of Protein Kinase CK2 Uncouples Bid Cleavage from Caspase-8 Activation. J. Cell Sci. 2010, 123, 1401–1406. [Google Scholar] [CrossRef]
- Berberich, S.J.; Cole, M.D. Casein Kinase II Inhibits the DNA-Binding Activity of Max Homodimers but Not Myc/Max Heterodimers. Genes Dev. 1992, 6, 166–176. [Google Scholar] [CrossRef]
- Bousset, K.; Henriksson, M.; Luscher-Firzlaff, J.M.; Litchfield, D.W.; Luscher, B. Identification of Casein Kinase II Phosphorylation Sites in Max: Effects on DNA-Binding Kinetics of Max Homo- and Myc/Max Heterodimers. Oncogene 1993, 8, 3211–3220. [Google Scholar]
- Krippner-Heidenreich, A.; Talanian, R.V.; Sekul, R.; Kraft, R.; Tholer, H.; Ottleben, H.; Lu, B. Targeting of the Transcription Factor Max during Apoptosis: Phosphorylation-Regulated Cleavage by Caspase-5 at an Unusual Glutamic Acid Residue in Position P1. Biochem. J. 2001, 358, 705–715. [Google Scholar] [CrossRef]
- Barrio Garcia, S.; Ruiz-Heredia, Y.; Da Via’, M.; Gallardo, M.; Garitano-Trojaola, A.; Zovko, J.; Raab, M.S.; Sonneveld, P.; Braggio, E.; Stewart, A.K.; et al. Role of MAX As a Tumor Suppressor Driver Gene in Multiple Myeloma. Blood 2017, 130, 4347. [Google Scholar] [CrossRef]
- Augert, A.; Mathsyaraja, H.; Ibrahim, A.H.; Freie, B.; Geuenich, M.J.; Cheng, P.F.; Alibeckoff, S.P.; Wu, N.; Hiatt, J.B.; Basom, R.; et al. MAX Functions as a Tumor Suppressor and Rewires Metabolism in Small Cell Lung Cancer. Cancer Cell 2020, 38, 97–114.e7. [Google Scholar] [CrossRef]
- Fan, Y.; Lu, D. The Ikaros Family of Zinc-Finger Proteins. Acta Pharm. Sin. B 2016, 6, 513. [Google Scholar] [CrossRef]
- Gurel, Z.; Ronni, T.; Ho, S.; Kuchar, J.; Payne, K.J.; Turk, C.W.; Dovat, S. Recruitment of Ikaros to Pericentromeric Heterochromatin Is Regulated by Phosphorylation. J. Biol. Chem. 2008, 283, 8291. [Google Scholar] [CrossRef]
- Popescu, M.; Gurel, Z.; Ronni, T.; Song, C.; Hung, K.Y.; Payne, K.J.; Dovat, S. Ikaros Stability and Pericentromeric Localization Are Regulated by Protein Phosphatase 1. J. Biol. Chem. 2009, 284, 13869. [Google Scholar] [CrossRef]
- Song, C.; Ge, Z.; Ding, Y.; Tan, B.H.; Desai, D.; Gowda, K.; Amin, S.; Gowda, R.; Robertson, G.P.; Yue, F.; et al. IKAROS and CK2 Regulate Expression of BCL-XL and Chemosensitivity in High-Risk B-Cell Acute Lymphoblastic Leukemia. Blood 2020, 136, 1520–1534. [Google Scholar] [CrossRef]
- Yat Ming Yung, B. Oncogenic Role of Nucleophosmin/B23. Chang Gung Med. J. 2007. [Google Scholar]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From Structure and Function to Disease Development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef]
- Negi, S.S.; Olson, M.O.J. Effects of Interphase and Mitotic Phosphorylation on the Mobility and Location of Nucleolar Protein B23. J. Cell Sci. 2006, 119, 3676–3685. [Google Scholar] [CrossRef]
- Szebeni, A.; Hingorani, I.; Negi, S.; Olson, M.O.J. Role of Protein Kinase CK2 Phosphorylation in the Molecular Chaperone Activity of Nucleolar Protein B23. J. Biol. Chem. 2003, 278, 9107–9115. [Google Scholar] [CrossRef]
- Wang, G.; Pan, Y.; Ahmad, K.A.; Ahmed, K. Protein B23/Nucleophosmin/Numatrin Nuclear Dynamics in Relation to Protein Kinase CK2 and Apoptotic Activity in Prostate Cells. Biochemistry 2010, 49, 3842–3852. [Google Scholar] [CrossRef]
- Perera, Y.; Farina, H.G.; Gil, J.; Rodriguez, A.; Benavent, F.; Castellanos, L.; Gómez, R.E.; Acevedo, B.E.; Alonso, D.F.; Perea, S.E. Anticancer Peptide CIGB-300 Binds to Nucleophosmin/B23, Impairs Its CK2-Mediated Phosphorylation, and Leads to Apoptosis through Its Nucleolar Disassembly Activity. Mol. Cancer Ther. 2009, 8, 1189–1196. [Google Scholar] [CrossRef]
- Perera, Y.; Pedroso, S.; Borras-Hidalgo, O.; Vázquez, D.M.; Miranda, J.; Villareal, A.; Falcón, V.; Cruz, L.D.; Farinas, H.G.; Perea, S.E. Pharmacologic Inhibition of the CK2-Mediated Phosphorylation of B23/NPM in Cancer Cells Selectively Modulates Genes Related to Protein Synthesis, Energetic Metabolism, and Ribosomal Biogenesis. Mol. Cell. Biochem. 2015, 404, 103–112. [Google Scholar] [CrossRef]
- Feroz, W.; Sheikh, A.M.A. Exploring the Multiple Roles of Guardian of the Genome: P53. Egypt. J. Med. Hum. Genet. 2020, 21, 49. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of P53 in Cell Death and Human Cancers. Cancers 2011, 3, 994. [Google Scholar] [CrossRef]
- Shen, Y.; White, E. P53-Dependent Apoptosis Pathways. Adv. Cancer Res. 2001, 82, 55–84. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.P.; Kraiss, S.; Montenarh, M. Association of Casein Kinase II with Immunopurified P53. Oncogene 1991, 6, 877–884. [Google Scholar] [PubMed]
- Meek, D.W.; Simon, S.; Kikkawa, U.; Eckhart, W. The P53 Tumour Suppressor Protein Is Phosphorylated at Serine 389 by Casein Kinase II. EMBO J. 1990, 9, 3253–3260. [Google Scholar] [CrossRef] [PubMed]
- Prowald, A.; Schuster, N.; Montenarh, M. Regulation of the DNA Binding of P53 by Its Interaction with Protein Kinase CK2. FEBS Lett. 1997, 408, 99–104. [Google Scholar] [CrossRef]
- Schuster, N.; Prowald, A.; Schneider, E.; Scheidtmann, K.H.; Montenarh, M. Regulation of P53 Mediated Transactivation by the Beta-Subunit of Protein Kinase CK2. FEBS Lett. 1999, 447, 160–166. [Google Scholar] [CrossRef]
- Meek, D.W.; Cox, M. Induction and Activation of the P53 Pathway: A Role for the Protein Kinase CK2? Mol. Cell. Biochem. 2011, 356, 133–138. [Google Scholar] [CrossRef]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant P53 Exerts a Dominant Negative Effect by Preventing Wild-Type P53 from Binding to the Promoter of Its Target Genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef]
- Gillotin, S.; Yap, D.; Lu, X. Mutation at Ser392 Specifically Sensitizes Mutant P53H175 to Mdm2-Mediated Degradation. Cell Cycle 2010, 9, 1390–1398. [Google Scholar] [CrossRef]
- Chao, C.C.K. Mechanisms of P53 Degradation. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 438, 139–147. [Google Scholar] [CrossRef]
- Khoronenkova, S.V.; Dianova, I.I.; Ternette, N.; Kessler, B.M.; Parsons, J.L.; Dianov, G.L.D. ATM-Dependent Downregulation of USP7/HAUSP by PPM1G Activates P53 Response to DNA Damage. Mol. Cell 2012, 45, 801–813. [Google Scholar] [CrossRef]
- Zhou, J.; Qiao, X.; Xiao, L.; Sun, W.; Wang, L.; Li, H.; Wu, Y.; Ding, X.; Hu, X.; Zhou, C.; et al. Identification and Characterization of the Novel Protein CCDC106 That Interacts with P53 and Promotes Its Degradation. FEBS Lett. 2010, 584, 1085–1090. [Google Scholar] [CrossRef]
- Ning, Y.; Wang, C.; Liu, X.; Du, Y.; Liu, S.; Liu, K.; Zhou, J.; Zhou, C. CK2-Mediated CCDC106 Phosphorylation Is Required for P53 Degradation in Cancer Progression. J. Exp. Clin. Cancer Res. 2019, 38, 131. [Google Scholar] [CrossRef]
- Lu, H.; Yan, C.; Quan, X.X.; Yang, X.; Zhang, J.; Bian, Y.; Chen, Z.; Van Waes, C. CK2 Phosphorylates and Inhibits TAp73 Tumor Suppressor Function to Promote Expression of Cancer Stem Cell Genes and Phenotype in Head and Neck Cancer. Neoplasia 2014, 16, 789–800. [Google Scholar] [CrossRef]
- Lu, H.; Yang, X.; Duggal, P.; Allen, C.T.; Yan, B.; Cohen, J.; Nottingham, L.; Romano, R.-A.; Sinha, S.; King, K.E.; et al. TNF-α Promotes c-REL/ΔNp63α Interaction and TAp73 Dissociation from Key Genes That Mediate Growth Arrest and Apoptosis in Head and Neck Cancer. Cancer Res. 2011, 71, 6867–6877. [Google Scholar] [CrossRef]
- Nagata, E.; Luo, H.R.; Saiardi, A.; Bae, B.I.; Suzuki, N.; Snyder, S.H. Inositol Hexakisphosphate Kinase-2, a Physiologic Mediator of Cell Death. J. Biol. Chem. 2005, 280, 1634–1640. [Google Scholar] [CrossRef]
- Koldobskiy, M.A.; Chakraborty, A.; Werner, J.K.; Snowman, A.M.; Juluri, K.R.; Vandiver, M.S.; Kim, S.; Heletz, S.; Snyder, S.H. P53-Mediated Apoptosis Requires Inositol Hexakisphosphate Kinase-2. Proc. Natl. Acad. Sci. USA 2010, 107, 20947–20951. [Google Scholar] [CrossRef]
- Chakraborty, A.; Werner, J.K.; Koldobskiy, M.A.; Mustafa, A.K.; Juluri, K.R.; Pietropaoli, J.; Snowman, A.M.; Snyder, S.H. Casein Kinase-2 Mediates Cell Survival through Phosphorylation and Degradation of Inositol Hexakisphosphate Kinase-2. Proc. Natl. Acad. Sci. USA 2011, 108, 2205–2209. [Google Scholar] [CrossRef]
- Rao, F.; Cha, J.; Xu, J.; Xu, R.; Vandiver, M.S.; Tyagi, R.; Tokhunts, R.; Koldobskiy, M.A.; Fu, C.; Barrow, R.; et al. Inositol Pyrophosphates Mediate the DNA-PK/ATM-P53 Cell Death Pathway by Regulating CK2 Phosphorylation of Tti1/Tel2. Mol. Cell 2014, 54, 119–132. [Google Scholar] [CrossRef]
- Kang, H.; Jung, J.W.; Kim, M.K.; Chung, J.H. CK2 Is the Regulator of SIRT1 Substrate-Binding Affinity, Deacetylase Activity and Cellular Response to DNA-Damage. PLoS ONE 2009, 4, e6611. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.-I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. HSIR2 (SIRT1) Functions as an NAD-Dependent P53 Deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Lee, J.T.; Gu, W. SIRT1: Regulator of P53 Deacetylation. Genes Cancer 2013, 4, 112–117. [Google Scholar] [CrossRef]
- Jang, S.Y.; Kim, S.Y.; Bae, Y.S. P53 Deacetylation by SIRT1 Decreases during Protein Kinase CKII Downregulation-Mediated Cellular Senescence. FEBS Lett. 2011, 585, 3360–3366. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, Y.; Huang, Q.; Tang, K. SIRT6 Regulates the Proliferation and Apoptosis of Hepatocellular Carcinoma via the ERK1/2 Signaling Pathway. Mol. Med. Rep. 2019, 20, 1575–1582. [Google Scholar] [CrossRef]
- Van Meter, M.; Mao, Z.; Gorbunova, V.; Seluanov, A. SIRT6 Overexpression Induces Massive Apoptosis in Cancer Cells but Not in Normal Cells. Cell Cycle 2011, 10, 3153–3158. [Google Scholar] [CrossRef]
- Desantis, V.; Lamanuzzi, A.; Vacca, A. The Role of SIRT6 in Tumors. Haematologica 2018, 103, 1–4. [Google Scholar] [CrossRef]
- Bae, J.S.; Park, S.H.; Jamiyandorj, U.; Kim, K.M.; Noh, S.J.; Kim, J.R.; Park, H.J.; Kwon, K.S.; Jung, S.H.; Park, H.S.; et al. CK2α/CSNK2A1 Phosphorylates SIRT6 and Is Involved in the Progression of Breast Carcinoma and Predicts Shorter Survival of Diagnosed Patients. Am. J. Pathol. 2016, 186, 3297–3315. [Google Scholar] [CrossRef]
- Khamis Hussein, U.; Gamal Ahmed, A.; Song, Y.; Min Kim, K.; Jae Moon, Y.; Ahn, A.-R.; Sung Park, H.; Jin Ahn, S.; Park, S.-H.; Ryul Kim, J.; et al. CK2α/CSNK2A1 Induces Resistance to Doxorubicin through SIRT6-Mediated Activation of the DNA Damage Repair Pathway. Cells 2021, 10, 1770. [Google Scholar] [CrossRef]
- Wei, X.; Liu, F.; Jiang, X.; Xu, X.; Zhou, T.; Kang, C. YY1 Promotes Telomerase Activity and Laryngeal Squamous Cell Carcinoma Progression Through Impairment of GAS5-Mediated P53 Stability. Front. Oncol. 2021, 11, 2759. [Google Scholar] [CrossRef]
- Meliala, I.T.S.; Hosea, R.; Kasim, V.; Wu, S. The Biological Implications of Yin Yang 1 in the Hallmarks of Cancer. Theranostics 2020, 10, 4183–4200. [Google Scholar] [CrossRef]
- Grönroos, E.; Terentiev, A.A.; Punga, T.; Ericsson, J. YY1 Inhibits the Activation of the P53 Tumor Suppressor in Response to Genotoxic Stress. Proc. Natl. Acad. Sci. USA 2004, 101, 12165–12170. [Google Scholar] [CrossRef]
- Riman, S.; Rizkallah, R.; Kassardjian, A.; Alexander, K.E.; Lüscher, B.; Hurt, M.M. Phosphorylation of the Transcription Factor YY1 by CK2 Prevents Cleavage by Caspase 7 during Apoptosis. Mol. Cell. Biol. 2012, 32, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xu, R.; Huang, X.; Tang, Z.; Tian, Y.; Zhang, J.; Zheng, X. Deubiquitinating Enzyme OTUB1 Promotes Cancer Cell Immunosuppression via Preventing ER-Associated Degradation of Immune Checkpoint Protein PD-L1. Cell Death Differ. 2020, 28, 1773–1789. [Google Scholar] [CrossRef] [PubMed]
- Saldana, M.; Saldana, M.; VanderVorst, K.; Berg, A.L.; Lee, H.; Carraway, K.L., III. Endocrine-Related Cancer Otubain 1: A Cancer-Associated Deubiquitinase R1-R14 Otubain 1: A Non-Canonical Deubiquitinase with an Emerging Role in Cancer. Endocr. Relat. Cancer 2019, 26, R1–R14. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-X.; Dai, M.-S. Deubiquitinating Enzyme Regulation of the P53 Pathway: A Lesson from Otub1. World J. Biol. Chem. 2014, 5, 75. [Google Scholar] [CrossRef]
- Lee, B.S.; Kang, S.U.; Huang, M.; Kim, Y.S.; Lee, Y.S.; Park, J.Y.; Kim, C.H. OTUB1 Knockdown Promotes Apoptosis in Melanoma Cells by Upregulating TRAIL Expression. BMB Rep. 2021, 54, 608. [Google Scholar] [CrossRef]
- Herhaus, L.; Perez-Oliva, A.B.; Cozza, G.; Gourlay, R.; Weidlich, S.; Campbell, D.G.; Pinna, L.A.; Sapkota, G.P. Casein Kinase 2 (CK2) Phosphorylates the Deubiquitylase OTUB1 at Ser16 to Trigger Its Nuclear Localization. Sci. Signal. 2015, 8. [Google Scholar] [CrossRef]
- Brancolini, C.; Iuliano, L. Proteotoxic Stress and Cell Death in Cancer Cells. Cancers 2020, 12, 2385. [Google Scholar] [CrossRef]
- Vydra, N.; Toma, A.; Widlak, W. Pleiotropic Role of HSF1 in Neoplastic Transformation. Curr. Cancer Drug Targets 2014, 14, 144. [Google Scholar] [CrossRef]
- Soncin, F.; Zhang, X.; Chu, B.; Wang, X.; Asea, A.; Stevenson, M.A.; Sacks, D.B.; Calderwood, S.K. Transcriptional Activity and DNA Binding of Heat Shock Factor-1 Involve Phosphorylation on Threonine 142 by CK2. Biochem. Biophys. Res. Commun. 2003, 303, 700–706. [Google Scholar] [CrossRef]
- Lu, W.-C.; Omari, R.; Ray, H.; Wang, J.; Williams, I.; Jacobs, C.; Hockaden, N.; Bochman, M.L.; Carpenter, R.L. AKT1 Mediates Multiple Phosphorylation Events That Functionally Promote HSF1 Activation. FEBS J. 2022, 289, 3876–3893. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The Role of the Unfolded Protein Response in Cancer Progression: From Oncogenesis to Chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- Manni, S.; Brancalion, A.; Tubi, L.Q.; Colpo, A.; Pavan, L.; Cabrelle, A.; Ave, E.; Zaffino, F.; Di Maira, G.; Ruzzene, M.; et al. Protein Kinase CK2 Protects Multiple Myeloma Cells from ER Stress-Induced Apoptosis and from the Cytotoxic Effect of HSP90 Inhibition through Regulation of the Unfolded Protein Response. Clin. Cancer Res. 2012, 18, 1888–1900. [Google Scholar] [CrossRef]
- Manni, S.; Carrino, M.; Piazza, F. Role of Protein Kinases CK1α and CK2 in Multiple Myeloma: Regulation of Pivotal Survival and Stress-Managing Pathways. J. Hematol. Oncol. 2017, 10, 157. [Google Scholar] [CrossRef]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; et al. Cytotoxic Activity of the Casein Kinase 2 Inhibitor CX-4945 against T-Cell Acute Lymphoblastic Leukemia: Targeting the Unfolded Protein Response Signaling. Leukemia 2013, 28, 543–553. [Google Scholar] [CrossRef]
- Hessenauer, A.; Schneider, C.C.; Götz Claudia, C.; Montenarh, M. CK2 Inhibition Induces Apoptosis via the ER Stress Response. Cell. Signal. 2011, 23, 145–151. [Google Scholar] [CrossRef]
- Pällmann, N.; Livgård, M.; Tesikova, M.; Zeynep Nenseth, H.; Akkus, E.; Sikkeland, J.; Jin, Y.; Koc, D.; Kuzu, O.F.; Pradhan, M.; et al. Regulation of the Unfolded Protein Response through ATF4 and FAM129A in Prostate Cancer. Oncogene 2019, 38, 6301–6318. [Google Scholar] [CrossRef]
- Ampofo, E.; Sokolowsky, T.; Götz, C.; Montenarh, M. Functional Interaction of Protein Kinase CK2 and Activating Transcription Factor 4 (ATF4), a Key Player in the Cellular Stress Response. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 439–451. [Google Scholar] [CrossRef]
- Sheshadri, N.; Poria, D.K.; Sharan, S.; Hu, Y.; Yan, C.; Koparde, V.N.; Balamurugan, K.; Sterneck, E. PERK Signaling through C/EBPδ Contributes to ER Stress-Induced Expression of Immunomodulatory and Tumor Promoting Chemokines by Cancer Cells. Cell Death Dis. 2021, 12, 1038. [Google Scholar] [CrossRef]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 10, 3083. [Google Scholar] [CrossRef]
- Schwind, L.; Zimmer, A.D.; Götz, C.; Montenarh, M. CK2 Phosphorylation of C/EBPδ Regulates Its Transcription Factor Activity. Int. J. Biochem. Cell Biol. 2015, 61, 81–89. [Google Scholar] [CrossRef]
- Ubeda, M.; Habener, J.F. CHOP Transcription Factor Phosphorylation by Casein Kinase 2 Inhibits Transcriptional Activation. J. Biol. Chem. 2003, 278, 40514–40520. [Google Scholar] [CrossRef]
- Schmitt, B.M.; Ampofo, E.; Stumpf, H.; Montenarh, M.; Götz, C. The Stability of CREB3/Luman Is Regulated by Protein Kinase CK2 Phosphorylation. Biochem. Biophys. Res. Commun. 2020, 523, 639–644. [Google Scholar] [CrossRef]
- Suh, D.Y. Review: Understanding Angiogenesis Its Clinical Applications. Ann. Clin. Lab. Sci. 2000, 30, 227–238. [Google Scholar]
- Schmitt, B.M.; Boewe, A.S.; Becker, V.; Nalbach, L.; Gu, Y.; Götz, C.; Menger, M.D.; Laschke, M.W.; Ampofo, E. Protein Kinase CK2 Regulates Nerve/Glial Antigen (NG)2-Mediated Angiogenic Activity of Human Pericytes. Cells 2020, 9, 1546. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Crivellato, E. The Role of Pericytes in Angiogenesis. Int. J. Dev. Biol. 2011, 55, 261–268. [Google Scholar] [CrossRef]
- Ljubimov, A.V.; Caballero, S.; Aoki, A.M.; Pinna, L.A.; Grant, M.B.; Castellon, R. Involvement of Protein Kinase CK2 in Angiogenesis and Retinal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4583–4591. [Google Scholar] [CrossRef]
- Kramerov, A.A.; Saghizadeh, M.; Caballero, S.; Shaw, L.C.; Li Calzi, S.; Bretner, M.; Montenarh, M.; Pinna, L.A.; Grant, M.B.; Ljubimov, A.V. Inhibition of Protein Kinase CK2 Suppresses Angiogenesis and Hematopoietic Stem Cell Recruitment to Retinal Neovascularization Sites. Mol. Cell. Biochem. 2008, 316, 177–186. [Google Scholar] [CrossRef]
- Pollreisz, A.; Afonyushkin, T.; Oskolkova, O.V.; Gruber, F.; Bochkov, V.N.; Schmidt-Erfurth, U. Retinal Pigment Epithelium Cells Produce VEGF in Response to Oxidized Phospholipids through Mechanisms Involving ATF4 and Protein Kinase CK2. Exp. Eye Res. 2013, 116, 177–184. [Google Scholar] [CrossRef]
- Feng, D.; Welker, S.; Körbel, C.; Rudzitis-Auth, J.; Menger, M.D.; Montenarh, M.; Laschke, M.W. Protein Kinase CK2 Is a Regulator of Angiogenesis in Endometriotic Lesions. Angiogenesis 2012, 15, 243–252. [Google Scholar] [CrossRef]
- Nagy, J.A.; Laura, A.E.; Ae, B.; Zeng, H.; Ann, A.E.; Dvorak, M.; Harold, A.E.; Dvorak, F. Vascular Permeability, Vascular Hyperpermeability and Angiogenesis. Angiogenesis 2008, 11, 109–119. [Google Scholar] [CrossRef]
- Deng, X.; Szabo, S.; Khomenko, T.; Tolstanova, G.; Paunovic, B.; French, S.W.; Sandor, Z. Novel Pharmacologic Approaches to the Prevention and Treatment of Ulcerative Colitis. Curr. Pharm. Des. 2012, 19, 17–28. [Google Scholar] [CrossRef]
- Irby, R.B.; Yeatman, T.J. Role of Src Expression and Activation in Human Cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Alimonti, A.; Scaglioni, P.P.; Koutcher, J.A.; Cordon-Cardo, C.; Pandolfi, P.P.; Experiments, P.P.P. Identification of a Tumour Suppressor Network Opposing Nuclear Akt Function. Nature 2006, 441, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Spring, K.; Lapointe, L.; Caron, C.; Langlois, S.; Royal, I. Phosphorylation of DEP-1/PTPRJ on Threonine 1318 Regulates Src Activation and Endothelial Cell Permeability Induced by Vascular Endothelial Growth Factor. Cell. Signal. 2014, 26, 1283–1293. [Google Scholar] [CrossRef]
- Chen, R.H.; Lee, Y.R.; Yuan, W.C. The Role of PML Ubiquitination in Human Malignancies. J. Biomed. Sci. 2012, 19, 81. [Google Scholar] [CrossRef]
- Bernardi, R.; Guernah, I.; Jin, D.; Grisendi, S.; Alimonti, A.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Celeste Simon, M.; Rafii, S.; Pandolfi, P.P. PML Inhibits HIF-1α Translation and Neoangiogenesis through Repression of MTOR. Nature 2006, 442, 779–785. [Google Scholar] [CrossRef]
- Scaglioni, P.P.; Yung, T.M.; Choi, S.C.; Baldini, C.; Konstantinidou, G.; Pandolfi, P.P. CK2 Mediates Phosphorylation and Ubiquitin-Mediated Degradation of the PML Tumor Suppressor. Mol. Cell. Biochem. 2008, 316, 149–154. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; de Thé, H. CK2 and PML: Regulating the Regulator. Cell 2006, 126, 244–245. [Google Scholar] [CrossRef]
- Mottet, D.; Ruys, S.P.D.; Demazy, C.; Raes, M.; Michiels, C. Role for Casein Kinase 2 in the Regulation of HIF-1 Activity. Int. J. Cancer 2005, 117, 764–774. [Google Scholar] [CrossRef]
- Semenza, G.L. Evaluation of HIF-1 Inhibitors as Anticancer Agents. Drug Discov. Today 2007, 12, 853–859. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.-H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of Vascular Endothelial Growth Factor Gene Transcription by Hypoxia-Inducible Factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Hu, K.; Babapoor-Farrokhran, S.; Rodrigues, M.; Deshpande, M.; Puchner, B.; Kashiwabuchi, F.; Junaid Hassan, S.; Asnaghi, L.; Handa, J.T.; Merbs, S.; et al. Hypoxia-Inducible Factor 1 Upregulation of Both VEGF and ANGPTL4 Is Required to Promote the Angiogenic Phenotype in Uveal Melanoma. Oncotarget 2016, 7, 7816–7828. [Google Scholar] [CrossRef]
- Haase, V.H. The VHL Tumor Suppressor: Master Regulator of HIF. Curr. Pharm. Des. 2009, 15, 3895–3903. [Google Scholar] [CrossRef]
- Pluemsampant, S.; Safronova, O.S.; Nakahama, K.I.; Morita, I. Protein Kinase CK2 Is a Key Activator of Histone Deacetylase in Hypoxia-Associated Tumors. Int. J. Cancer 2008, 122, 333–341. [Google Scholar] [CrossRef]
- Ampofo, E.; Kietzmann, T.; Zimmer, A.; Jakupovic, M.; Montenarh, M.; Götz, C. Phosphorylation of the von Hippel-Lindau Protein (VHL) by Protein Kinase CK2 Reduces Its Protein Stability and Affects P53 and HIF-1α Mediated Transcription. Int. J. Biochem. Cell Biol. 2010, 42, 1729–1735. [Google Scholar] [CrossRef]
- Hubert, A.; Paris, S.; Piret, J.P.; Ninane, N.; Raes, M.; Michiels, C. Casein Kinase 2 Inhibition Decreases Hypoxia-Inducible Factor-1 Activity under Hypoxia through Elevated P53 Protein Level. J. Cell Sci. 2006, 119, 3351–3362. [Google Scholar] [CrossRef]
- Guerra, B.; Rasmussen, T.D.L.; Schnitzler, A.; Jensen, H.H.; Boldyreff, B.S.; Miyata, Y.; Marcussen, N.; Niefind, K.; Issinger, O.G. Protein Kinase CK2 Inhibition Is Associated with the Destabilization of HIF-1α in Human Cancer Cells. Cancer Lett. 2015, 356, 751–761. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.-J.; Mimnaugh, E.G.; Martinez, A.; Cuttitta, F.; Neckers, L.M. Hsp90 Regulates a von Hippel Lindau-Independent Hypoxia-Inducible Factor-1 Alpha-Degradative Pathway. J. Biol. Chem. 2002, 277, 29936–29944. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. The Biology of Cancer Metastasis: Historical Perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef]
- Deshiere, A.; Duchemin-Pelletier, E.; Spreux, E.; Ciais, D.; Combes, F.; Vandenbrouck, Y.; Couté, Y.; Mikaelian, I.; Giusiano, S.; Charpin, C.; et al. Unbalanced Expression of CK2 Kinase Subunits Is Sufficient to Drive Epithelial-to-Mesenchymal Transition by Snail1 Induction. Oncogene 2013, 32, 1373–1383. [Google Scholar] [CrossRef]
- Giusiano, S.; Cochet, C.; Filhol, O.; Duchemin-Pelletier, E.; Secq, V.; Bonnier, P.; Carcopino, X.; Boubli, L.; Birnbaum, D.; Garcia, S.; et al. Protein Kinase CK2α Subunit Over-Expression Correlates with Metastatic Risk in Breast Carcinomas: Quantitative Immunohistochemistry in Tissue Microarrays. Eur. J. Cancer 2011, 47, 792–801. [Google Scholar] [CrossRef]
- Montenarh, M. Protein Kinase CK2 and Angiogenesis. Adv. Clin. Exp. Med. 2014, 23, 153–158. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an Orally Bioavailable Selective Inhibitor of Protein Kinase CK2, Inhibits Prosurvival and Angiogenic Signaling and Exhibits Antitumor Efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef]
- Graziano, F. The E-Cadherin Gene, Structure and Function. Spotlight Fam. Hered. Gastric Cancer 2013, 13, 27–33. [Google Scholar] [CrossRef]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-Cadherin in Cancer Metastasis, Its Emerging Role in Haematological Malignancies and Potential as a Therapeutic Target in Cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Zou, J.; Luo, H.; Zeng, Q.; Dong, Z.; Wu, D.; Liu, L. Protein Kinase CK2α Is Overexpressed in Colorectal Cancer and Modulates Cell Proliferation and Invasion via Regulating EMT-Related Genes. J. Transl. Med. 2011, 9, 97. [Google Scholar] [CrossRef]
- Ko, H.; Kim, S.; Jin, C.H.; Lee, E.; Ham, S.; Yook, J.I.; Kim, K. Protein Kinase Casein Kinase 2-Mediated Upregulation of N-Cadherin Confers Anoikis Resistance on Esophageal Carcinoma Cells. Mol. Cancer Res. MCR 2012, 10, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.; Kim, S.; Yang, K.; Kim, K. Phosphorylation-Dependent Stabilization of MZF1 Upregulates N-Cadherin Expression during Protein Kinase CK2-Mediated Epithelial-Mesenchymal Transition. Oncogenesis 2018, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Abiatari, I.; Gillen, S.; DeOliveira, T.; Klose, T.; Bo, K.; Giese, N.A.; Friess, H.; Kleeff, J. The Microtubule-Associated Protein MAPRE2 Is Involved in Perineural Invasion of Pancreatic Cancer Cells. Int. J. Oncol. 2009, 35, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Stenner, F.; Liewen, H.; Göttig, S.; Henschler, R.; Markuly, N.; Kleber, S.; Faust, M.; Mischo, A.; Bauer, S.; Zweifel, M.; et al. RP1 Is a Phosphorylation Target of CK2 and Is Involved in Cell Adhesion. PLoS ONE 2013, 8, e67595. [Google Scholar] [CrossRef] [PubMed]
- Seger, D.; Gechtman, Z.; Shaltiel, S. Phosphorylation of Vitronectin by Casein Kinase II Identification of the Sites and Their Promotion of Cell Adhesion and Spreading. J. Biol. Chem. 1998, 273, 24805–24813. [Google Scholar] [CrossRef]
- Seger, D.; Seger, R.; Shaltiel, S. The CK2 Phosphorylation of Vitronectin Promotion of Cell Adhesion via the V 3-Phosphatidylinositol 3-Kinase Pathway. J. Biol. Chem. 2001, 276, 16998–17006. [Google Scholar] [CrossRef]
- Martin, T.A.; Jiang, W.G. Loss of Tight Junction Barrier Function and Its Role in Cancer Metastasis. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 872–891. [Google Scholar] [CrossRef]
- Wang, M.; Liu, Y.; Qian, X.; Wei, N.; Tang, Y.; Yang, J. Downregulation of Occludin Affects the Proliferation, Apoptosis and Metastatic Properties of Human Lung Carcinoma. Oncol. Rep. 2018, 40, 454–462. [Google Scholar] [CrossRef]
- Martin, T.A.; Mansel, R.E.; Jiang, W.G. Loss of Occludin Leads to the Progression of Human Breast Cancer. Int. J. Mol. Med. 2010, 26, 723–734. [Google Scholar] [CrossRef]
- Metastasis to Bone in Human Cancer Is Associated with Loss of Occludin Expression|Anticancer Research. Available online: https://ar.iiarjournals.org/content/36/3/1287 (accessed on 5 May 2022).
- Raleigh, D.R.; Boe, D.M.; Yu, D.; Weber, C.R.; Marchiando, A.M.; Bradford, E.M.; Wang, Y.; Wu, L.; Schneeberger, E.E.; Shen, L.; et al. Occludin S408 Phosphorylation Regulates Tight Junction Protein Interactions and Barrier Function. J. Cell Biol. 2011, 193, 565–582. [Google Scholar] [CrossRef]
- Dörfel, M.J.; Westphal, J.K.; Bellmann, C.; Krug, S.M.; Cording, J.; Mittag, S.; Tauber, R.; Fromm, M.; Blasig, I.E.; Huber, O. CK2-Dependent Phosphorylation of Occludin Regulates the Interaction with ZO-Proteins and Tight Junction Integrity. Cell Commun. Signal. 2013, 11, 40. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Ebright, R.Y.; Haber, D.A.; Maheswaran, S. Translational Regulation of Cancer Metastasis. Cancer Res. 2021, 81, 517–524. [Google Scholar] [CrossRef]
- Konicek, B.W.; Dumstorf, C.A.; Graff, J.R. Targeting the EIF4F Translation Initiation Complex for Cancer Therapy. Cell Cycle 2008, 7, 2466–2471. [Google Scholar] [CrossRef]
- Ye, Q.; Cai, W.; Zheng, Y.; Evers, B.M.; She, Q.B. ERK and AKT Signaling Cooperate to Translationally Regulate Survivin Expression for Metastatic Progression of Colorectal Cancer. Oncogene 2014, 33, 1828–1839. [Google Scholar] [CrossRef]
- Gandin, V.; Masvidal, L.; Cargnello, M.; Gyenis, L.; McLaughlan, S.; Cai, Y.; Tenkerian, C.; Morita, M.; Balanathan, P.; Jean-Jean, O.; et al. MTORC1 and CK2 Coordinate Ternary and EIF4F Complex Assembly. Nat. Commun. 2016, 7, 11127. [Google Scholar] [CrossRef]
- Schevzov, G.; Kee, A.J.; Wang, B.; Sequeira, V.B.; Hook, J.; Coombes, J.D.; Lucas, C.A.; Stehn, J.R.; Musgrove, E.A.; Cretu, A.; et al. Regulation of Cell Proliferation by ERK and Signal-Dependent Nuclear Translocation of ERK Is Dependent on Tm5NM1-Containing Actin Filaments. Mol. Biol. Cell 2015, 26, 2475–2490. [Google Scholar] [CrossRef]
- Angermayr, M.; Hochleitner, E.; Lottspeich, F.; Bandlow, W. Protein Kinase CK2 Activates the Atypical Rio1p Kinase and Promotes Its Cell-Cycle Phase-Dependent Degradation in Yeast. Authors J. Compil. 2007, 274, 4654–4667. [Google Scholar] [CrossRef]
- Kubiński, K.; Masłyk, M. The Link between Protein Kinase CK2 and Atypical Kinase Rio1. Pharmaceuticals 2017, 10, 21. [Google Scholar] [CrossRef]
- Weinberg, F.; Reischmann, N.; Fauth, L.; Taromi, S.; Mastroianni, J.; Köhler, M.; Halbach, S.; Becker, A.C.; Deng, N.; Schmitz, T.; et al. The Atypical Kinase RIOK1 Promotes Tumor Growth and Invasive Behavior. eBioMedicine 2017, 20, 79–97. [Google Scholar] [CrossRef]
- Hong, X.; Huang, H.; Qiu, X.; Ding, Z.; Feng, X.; Zhu, Y.; Zhuo, H.; Hou, J.; Zhao, J.; Cai, W.; et al. Targeting Posttranslational Modifications of RIOK1 Inhibits the Progression of Colorectal and Gastric Cancers. eLife 2018, 7. [Google Scholar] [CrossRef]
- Wen, S.; Niu, Y.; Huang, H. Posttranslational Regulation of Androgen Dependent and Independent Androgen Receptor Activities in Prostate Cancer. Asian J. Urol. 2020, 7, 203–218. [Google Scholar] [CrossRef]
- Trembley, J.H.; Kren, B.T.; Abedin, M.J.; Shaughnessy, D.P.; Li, Y.; Dehm, S.M.; Ahmed, K. CK2 Pro-Survival Role in Prostate Cancer Is Mediated via Maintenance and Promotion of Androgen Receptor and NFκB P65 Expression. Pharmaceuticals 2019, 12, 89. [Google Scholar] [CrossRef]
- Zhu, M.-L.; Kyprianou, N. Role of Androgens and the Androgen Receptor in Epithelial-Mesenchymal Transition and Invasion of Prostate Cancer Cells. FASEB J. 2010, 24, 769. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, P.; Huang, H.; Ye, X.; Chen, W.; Xu, G.; Zhang, F. Androgen Receptor Regulates EIF5A2 Expression and Promotes Prostate Cancer Metastasis via EMT. Cell Death Discov. 2021, 7, 373. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Lim, B.J.; Choi, H.K.; Hong, S.W.; Jang, H.S.; Kim, C.; Chun, K.H.; Choi, K.C.; Yoon, H.G. CK2-NCoR Signaling Cascade Promotes Prostate Tumorigenesis. Oncotarget 2013, 4, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Choi, H.K.; Choi, K.C.; Park, S.Y.; Ota, I.; Yook, J.I.; Lee, Y.H.; Kim, K.; Yoon, H.G. Nuclear Hormone Receptor Corepressor Promotes Esophageal Cancer Cell Invasion by Transcriptional Repression of Interferon-γ-Inducible Protein 10 in a Casein Kinase 2-Dependent Manner. Mol. Biol. Cell 2012, 23, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Amin, E.B.; Mayo, M.W.; Chudgar, N.P.; Bucciarelli, P.R.; Kadota, K.; Adusumilli, P.S.; Jones, D.R. CK2α’ Drives Lung Cancer Metastasis by Targeting BRMS1 Nuclear Export and Degradation. Cancer Res. 2016, 76, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Burns, J.A.; Cheney, C.A.; Zhang, N.; Vitelli, S.; Wang, F.; Bett, A.; Chastain, M.; Audoly, L.P.; Zhang, Z.-Q. Distinct Expression Profiles of Notch-1 Protein in Human Solid Tumors: Implications for Development of Targeted Therapeutic Monoclonal Antibodies. Biol. Targets Ther. 2010, 4, 163. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Ranganathan, P.; Vasquez-Del Carpio, R.; Kaplan, F.M.; Wang, H.; Gupta, A.; VanWye, J.D.; Capobianco, A.J. Hierarchical Phosphorylation within the Ankyrin Repeat Domain Defines a Phosphoregulatory Loop That Regulates Notch Transcriptional Activity. J. Biol. Chem. 2011, 286, 28844–28857. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 Signaling Regulates the Epithelial-Mesenchymal Transition and Invasion of Breast Cancer in a Slug-Dependent Manner. Mol. Cancer 2015, 14, 1–17. [Google Scholar] [CrossRef]
- Zhang, X.S.; Hu, Y.H.; Gao, H.Y.; Lan, X.W.; Xue, Y.W. Downregulation of Notch1 Inhibits the Invasion and Metastasis of Human Gastric Cancer Cells SGC7901 and MKN74 in Vitro through PTEN Activation and Dephosphorylation of Akt and FAK. Mol. Med. Rep. 2017, 16, 2318–2324. [Google Scholar] [CrossRef]
- Hu, Y.J.; Li, H.Y.; Qiu, K.J.; Li, D.C.; Zhou, J.H.; Hu, Y.H.; Zhang, F.M. Downregulation of Notch1 Inhibits the Invasion of Human Hepatocellular Carcinoma HepG2 and MHCC97H Cells through the Regulation of PTEN and FAK. Int. J. Mol. Med. 2014, 34, 1081–1086. [Google Scholar] [CrossRef]
- Zhang, S.; Long, H.; Yang, Y.L.; Wang, Y.; Hsieh, D.; Li, W.; Au, A.; Stoppler, H.J.; Xu, Z.; Jablons, D.M.; et al. Inhibition of CK2α Down-Regulates Notch1 Signalling in Lung Cancer Cells. J. Cell. Mol. Med. 2013, 17, 854–862. [Google Scholar] [CrossRef]
- Lian, H.; Li, D.; Zhou, Y.; Landesman-Bollag, E.; Zhang, G.; Anderson, N.M.; Tang, K.C.; Roderick, J.E.; Kelliher, M.A.; Seldin, D.C.; et al. CK2 Inhibitor CX-4945 Destabilizes NOTCH1 and Synergizes with JQ1 against Human T-Acute Lymphoblastic Leukemic Cells. Haematologica 2017, 102, e17–e21. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; De Gramont, A. Targeting the TGFβ Pathway for Cancer Therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef]
- Kim, S.; Ham, S.; Yang, K.; Kim, K. Protein Kinase CK2 Activation Is Required for Transforming Growth Factor β-Induced Epithelial–Mesenchymal Transition. Mol. Oncol. 2018, 12, 1811–1826. [Google Scholar] [CrossRef]
- The Regulation of PRH/HHEX by Transforming Growth Factor. Available online: http://research-information.bristol.ac.uk (accessed on 5 May 2022).
- The Role of PRH/HHEX in TGF-β Signaling and Cancer Cell-Platelet Interactions—University of Bristol. Available online: https://research-information.bris.ac.uk/en/studentTheses/the-role-of-prhhhex-in-tgf-β-signaling-and-cancer-cell-platelet-i (accessed on 3 May 2022).
- Ji, H.; Wang, J.; Nika, H.; Hawke, D.; Keezer, S.; Ge, Q.; Fang, B.; Fang, X.; Fang, D.; Litchfield, D.W.; et al. EGF-Induced ERK Activation Promotes CK2-Mediated Disassociation of α-Catenin from β-Catenin and Transactivation of β-Catenin. Mol. Cell 2009, 36, 547–559. [Google Scholar] [CrossRef]
- Kim, W.K.; Kwon, Y.; Jang, M.; Park, M.; Kim, J.; Cho, S.; Jang, D.G.; Lee, W.B.; Jung, S.H.; Choi, H.J.; et al. β-Catenin Activation down-Regulates Cell-Cell Junction-Related Genes and Induces Epithelial-to-Mesenchymal Transition in Colorectal Cancers. Sci. Rep. 2019, 9, 18440. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; De Barrios, O.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. β-Catenin/TCF4 Complex Induces the Epithelial-to-Mesenchymal Transition (EMT)-Activator ZEB1 to Regulate Tumor Invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209. [Google Scholar] [CrossRef]
- Du, L.; Lee, J.-H.; Jiang, H.; Wang, C.; Wang, S.; Zheng, Z.; Shao, F.; Xu, D.; Xia, Y.; Li, J.; et al. β-Catenin Induces Transcriptional Expression of PD-L1 to Promote Glioblastoma Immune Evasion. J. Exp. Med. 2020, 217, e20191115. [Google Scholar] [CrossRef]
- Eichberger, J.; Schulz, D.; Pscheidl, K.; Fiedler, M.; Reichert, T.E.; Bauer, R.J.; Ettl, T. PD-L1 Influences Cell Spreading, Migration and Invasion in Head and Neck Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8089. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, Y.; Chu, Y.-Y.; Li, Y.; Hsu, J.-M.; Jiang, Z.; Liu, C.; Hsu, J.L.; Chang, W.-C.; Yang, R.; et al. Phosphorylation and Stabilization of PD-L1 by CK2 Suppresses Dendritic Cell FunctionCK2 Phosphorylates PD-L1 to Inhibit Dendritic Cell Function. Cancer Res. 2022, 82, 2185–2195. [Google Scholar] [CrossRef]
- Tapia, J.C.; Niechi, I. Endothelin-Converting Enzyme-1 in Cancer Aggressiveness. Cancer Lett. 2019, 452, 152–157. [Google Scholar] [CrossRef]
- Pérez-Moreno, P.; Quezada-Meza, C.; Chavez-Almarza, C.; Niechi, I.; Silva-Pavez, E.; Trigo-Hidalgo, C.; Aguayo, F.; Jara, L.; Cáceres-Verschae, A.; Varas-Godoy, M.; et al. Phosphorylation of Endothelin-Converting Enzyme-1c at Serines 18 and 20 by CK2 Promotes Aggressiveness Traits in Colorectal Cancer Cells. Front. Oncol. 2020, 10, 1004. [Google Scholar] [CrossRef]
- Pérez-Moreno, P.; Indo, S.; Niechi, I.; Huerta, H.; Cabello, P.; Jara, L.; Aguayo, F.; Varas-Godoy, M.; Burzio, V.A.; Tapia, J.C. Endothelin-Converting Enzyme-1c Promotes Stem Cell Traits and Aggressiveness in Colorectal Cancer Cells. Mol. Oncol. 2020, 14, 347–362. [Google Scholar] [CrossRef]
- Niechi, I.; Silva, E.; Cabello, P.; Huerta, H.; Carrasco, V.; Villar, P.; Cataldo, L.R.; Marcelain, K.; Armisen, R.; Varas-Godoy, M.; et al. Colon Cancer Cell Invasion Is Promoted by Protein Kinase CK2 through Increase of Endothelin-Converting Enzyme-1c Protein Stability. Oncotarget 2015, 6, 42749. [Google Scholar] [CrossRef]
- Ninsontia, C.; Phiboonchaiyanan, P.P.; Chanvorachote, P. Zinc Induces Epithelial to Mesenchymal Transition in Human Lung Cancer H460 Cells via Superoxide Anion-Dependent Mechanism. Cancer Cell Int. 2016, 16, 48. [Google Scholar] [CrossRef]
- Manzo, G. Similarities Between Embryo Development and Cancer Process Suggest New Strategies for Research and Therapy of Tumors: A New Point of View. Front. Cell Dev. Biol. 2019, 7, 20. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8. [Google Scholar] [CrossRef]
- Di Magliano, M.P.; Hebrok, M. Hedgehog Signalling in Cancer Formation and Maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef]
- Avery, J.T.; Zhang, R.; Boohaker, R.J. GLI1: A Therapeutic Target for Cancer. Front. Oncol. 2021, 11, 1833. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, J.; Hu, Y.; Lu, P.; Luo, Q.; Wang, L. Gli Promotes Tumor Progression through Regulating Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. J. Cardiothorac. Surg. 2020, 15, 18. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Mao, J.H.; Hsieh, D.; Kim, I.J.; Hu, L.M.; Xu, Z.; Long, H.; Jablons, D.M.; You, L. Inhibition of CK2α Down-Regulates Hedgehog/Gli Signaling Leading to a Reduction of a Stem-Like Side Population in Human Lung Cancer Cells. PLoS ONE 2012, 7, e38996. [Google Scholar] [CrossRef]
- Wu, D.; Sui, C.; Meng, F.; Tian, X.; Fu, L.; Li, Y.; Qi, X.; Cui, H.; Liu, Y.; Jiang, Y. Stable Knockdown of Protein Kinase CK2-Alpha (CK2α) Inhibits Migration and Invasion and Induces Inactivation of Hedgehog Signaling Pathway in Hepatocellular Carcinoma Hep G2 Cells. Acta Histochem. 2014, 116, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, Y.L.; Wang, Y.; You, B.; Dai, Y.; Chan, G.; Hsieh, D.; Kim, I.J.; Fang, L.T.; Au, A.; et al. CK2α, over-Expressed in Human Malignant Pleural Mesothelioma, Regulates the Hedgehog Signaling Pathway in Mesothelioma Cells. J. Exp. Clin. Cancer Res. CR 2014, 33, 93. [Google Scholar] [CrossRef] [PubMed]
- Purzner, T.; Purzner, J.; Buckstaff, T.; Cozza, G.; Gholamin, S.; Rusert, J.M.; Hartl, T.A.; Sanders, J.; Conley, N.; Ge, X.; et al. Developmental Phosphoproteomics Identifies the Kinase CK2 as a Driver of Hedgehog Signaling and a Therapeutic Target in Medulloblastoma. Sci. Signal. 2018, 11, eaau5147. [Google Scholar] [CrossRef]
- Tang, H.; Massi, D.; Hemmings, B.A.; Mandalà, M.; Hu, Z.; Wicki, A.; Xue, G. AKT-Ions with a TWIST between EMT and MET. Oncotarget 2016, 7, 62767–62777. [Google Scholar] [CrossRef]
- Su, Y.W.; Xie, T.X.; Sano, D.; Myers, J.N. IL-6 Stabilizes Twist and Enhances Tumor Cell Motility in Head and Neck Cancer Cells through Activation of Casein Kinase 2. PLoS ONE 2011, 6, e19412. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of Extracellular Matrix Remodelling in Tumour Progression and Metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Efthymiou, G.; Saint, A.; Ruff, M.; Rekad, Z.; Ciais, D.; Van Obberghen-Schilling, E. Shaping Up the Tumor Microenvironment with Cellular Fibronectin. Front. Oncol. 2020, 10, 641. [Google Scholar] [CrossRef]
- Yalak, G.; Shiu, J.Y.; Schoen, I.; Mitsi, M.; Vogel, V. Phosphorylated Fibronectin Enhances Cell Attachment and Upregulates Mechanical Cell Functions. PLoS ONE 2019, 14, e0218893. [Google Scholar] [CrossRef]
- Du, M.; Liu, J.; Chen, X.; Xie, Y.; Yuan, C.; Xiang, Y.; Sun, B.; Lan, K.; Chen, M.; James, S.J.; et al. Casein Kinase II Controls TBK1/IRF3 Activation in IFN Response against Viral Infection. J. Immunol. 2015, 194, 4477–4488. [Google Scholar] [CrossRef]
- Nora Pencheva, A.; de Gooijer, M.C.; Vis, D.J.; Wessels, L.F.; van Tellingen, O. Identification of a Druggable Pathway Controlling Glioblastoma Invasiveness. CellReports 2017, 20, 48–60. [Google Scholar] [CrossRef]
- Ku, M.J.; Park, J.W.; Ryu, B.J.; Son, Y.J.; Kim, S.H.; Lee, S.Y. CK2 Inhibitor CX4945 Induces Sequential Inactivation of Proteins in the Signaling Pathways Related with Cell Migration and Suppresses Metastasis of A549 Human Lung Cancer Cells. Bioorganic Med. Chem. Lett. 2013, 23, 5609–5613. [Google Scholar] [CrossRef]
- Guerra, B.; Siemer, S.; Boldyreff, B.; Issinger, O.G. Protein Kinase CK2: Evidence for a Protein Kinase CK2beta Subunit Fraction, Devoid of the Catalytic CK2alpha Subunit, in Mouse Brain and Testicles. FEBS Lett. 1999, 462, 353–357. [Google Scholar] [CrossRef]
- Pinna, L.A.; Meggio, F. Protein Kinase CK2 (“casein Kinase-2”) and Its Implication in Cell Division and Proliferation. Prog. Cell Cycle Res. 1997, 3, 77–97. [Google Scholar] [CrossRef]
- Stalter, G.; Siemer, S.; Becht, E.; Ziegler, M.; Remberger, K.; Issinger, O.G. Asymmetric Expression of Protein Kinase CK2 Subunits in Human Kidney Tumors. Biochem. Biophys. Res. Commun. 1994, 202, 141–147. [Google Scholar] [CrossRef]
- Domańska, K.; Zieliński, R.; Kubiński, K.; Sajnaga, E.; Masłyk, M.; Bretner, M.; Szyszka, R. Different Properties of Four Molecular Forms of Protein Kinase CK2 from Saccharomyces Cerevisiae. Acta Biochim. Pol. 2005, 52, 947–951. [Google Scholar] [CrossRef]
- Kaufhold, S.; Bonavida, B. Central Role of Snail1 in the Regulation of EMT and Resistance in Cancer: A Target for Therapeutic Intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef]
- Golden, D.; Cantley, L.G. Casein Kinase 2 Prevents Mesenchymal Transformation by Maintaining Foxc2 in the Cytoplasm. Oncogene 2015, 34, 4702–4712. [Google Scholar] [CrossRef]
- Filhol, O.; Giacosa, S.; Wallez, Y.; Cochet, C. Protein Kinase CK2 in Breast Cancer: The CK2β Regulatory Subunit Takes Center Stage in Epithelial Plasticity. Cell. Mol. Life Sci. 2015, 72, 3305–3322. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Weinhouse, S.; Warburg, O.; Burk, D.; Schade, A.L. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic Reprogramming by the PI3K-Akt-MTOR Pathway in Cancer. Recent Results Cancer Res. 2016, 207, 39–72. [Google Scholar] [CrossRef]
- Silva-Pavez, E.; Tapia, J. Protein Kinase CK2 in Cancer Energetics. Front. Oncol. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Im, D.K.; Cheong, H.; Lee, J.S.; Oh, M.K.; Yang, K.M. Protein Kinase CK2-Dependent Aerobic Glycolysis-Induced Lactate Dehydrogenase A Enhances the Migration and Invasion of Cancer Cells. Sci. Rep. 2019, 9, 5337. [Google Scholar] [CrossRef]
- Yang, K.M.; Kim, K. Protein Kinase CK2 Modulation of Pyruvate Kinase M Isoforms Augments the Warburg Effect in Cancer Cells. J. Cell. Biochem. 2018, 119, 8501–8510. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate Dehydrogenase A: A Key Player in Carcinogenesis and Potential Target in Cancer Therapy. Cancer Med. 2018, 7, 6124. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer Metabolism and the Warburg Effect: The Role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Ponce, D.P.; Maturana, J.L.; Cabello, P.; Yefi, R.; Niechi, I.; Silva, E.; Armisen, R.; Galindo, M.; Antonelli, M.; Tapia, J.C. Phosphorylation of AKT/PKB by CK2 Is Necessary for the AKT-Dependent up-Regulation of β-Catenin Transcriptional Activity. J. Cell. Physiol. 2011, 226, 1953–1959. [Google Scholar] [CrossRef]
- El-Sahli, S.; Xie, Y.; Wang, L.; Liu, S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers 2019, 11, 904. [Google Scholar] [CrossRef]
- Wu, H.; Symes, K.; Seldin, D.C.; Dominguez, I. Threonine 393 of β-Catenin Regulates Interaction with Axin. J. Cell. Biochem. 2009, 108, 52. [Google Scholar] [CrossRef]
- Ponce, D.P.; Yefi, R.; Cabello, P.; Maturana, J.L.; Niechi, I.; Silva, E.; Galindo, M.; Antonelli, M.; Marcelain, K.; Armisen, R.; et al. CK2 Functionally Interacts with AKT/PKB to Promote the β-Catenin-Dependent Expression of Survivin and Enhance Cell Survival. Mol. Cell. Biochem. 2011, 356, 127–132. [Google Scholar] [CrossRef]
- Rivadeneira, D.B.; Caino, M.C.; Seo, J.H.; Angelin, A.; Wallace, D.C.; Languino, L.R.; Altieri, D.C. Survivin Promotes Oxidative Phosphorylation, Subcellular Mitochondrial Repositioning, and Tumor Cell Invasion. Sci. Signal. 2015, 8, ra80. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.-N. Cancers The Key Role of the WNT/β-Catenin Pathway in Metabolic Reprogramming in Cancers under Normoxic Conditions. Cancers 2021, 13, 5557. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Zeng, K.W.; Wang, J.K.; Wang, L.C.; Guo, Q.; Liu, T.T.; Wang, F.J.; Feng, N.; Zhang, X.W.; Liao, L.X.; Zhao, M.M.; et al. Small Molecule Induces Mitochondrial Fusion for Neuroprotection via Targeting CK2 without Affecting Its Conventional Kinase Activity. Signal Transduct. Target. Ther. 2021, 6, 71. [Google Scholar] [CrossRef]
- Muhamad Hanif, I.; Pervaiz, S. Repressing the Activity of Protein Kinase CK2 Releases Mitochondria-Mediated Apoptosis in Cancer Cells. Curr. Drug Targets 2011, 12, 902–908. [Google Scholar] [CrossRef]
- Afzal, M.; Kren, B.T.; Naveed, A.K.; Trembley, J.H.; Ahmed, K. Protein Kinase CK2 Impact on Intracellular Calcium Homeostasis in Prostate Cancer. Mol. Cell. Biochem. 2020, 470, 131–143. [Google Scholar] [CrossRef]
- Dixit, D.; Ahmad, F.; Ghildiyal, R.; Joshi, S.D.; Sen, E. CK2 Inhibition Induced PDK4-AMPK Axis Regulates Metabolic Adaptation and Survival Responses in Glioma. Exp. Cell Res. 2016, 344, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Issinger, O.G. Role of Protein Kinase Ck2 in Aberrant Lipid Metabolism in Cancer. Pharmaceuticals 2020, 13, 292. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.; Levine, B.; Debnath, J. Autophagy and Cancer Metabolism. Methods Enzymol. 2014, 542, 25. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.B.; Svenstrup, T.H.; Guerra, B. Downregulation of Protein Kinase CK2 Induces Autophagic Cell Death through Modulation of the MTOR and MAPK Signaling Pathways in Human Glioblastoma Cells. Int. J. Oncol. 2012, 41, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-W.; Cheng, J.-J.; Huang, W.-Y.; Chang, Y.-F. Inhibition of CK2 by CX4945 to Induce Autophagy-Mediated Cell Death through Dephosphorylating Acetyl-CoA Carboxylase in Squamous Cell Carcinoma of Head and Neck Cancer. J. Clin. Oncol. 2017, 35, e17503. [Google Scholar] [CrossRef]
- Lee, S.W.; Song, Y.S.; Lee, S.Y.; Yoon, Y.G.; Lee, S.H.; Park, B.S.; Yun, I.; Choi, H.; Kim, K.; Chung, W.T.; et al. Downregulation of Protein Kinase CK2 Activity Facilitates Tumor Necrosis Factor-α-Mediated Chondrocyte Death through Apoptosis and Autophagy. PLoS ONE 2011, 6, e19163. [Google Scholar] [CrossRef]
- Silva-Pavez, E.; Villar, P.; Trigo, C.; Caamaño, E.; Niechi, I.; Pérez, P.; Muñoz, J.P.; Aguayo, F.; Burzio, V.A.; Varas-Godoy, M.; et al. CK2 Inhibition with Silmitasertib Promotes Methuosis-like Cell Death Associated to Catastrophic Massive Vacuolization of Colorectal Cancer Cells. Cell Death Dis. 2019, 10, 73. [Google Scholar] [CrossRef]
- Jang, D.E.; Song, J.; Park, J.W.; Yoon, S.H.; Bae, Y.S. Protein Kinase CK2 Activates Nrf2 via Autophagic Degradation of Keap1 and Activation of AMPK in Human Cancer Cells. BMB Rep. 2020, 53, 272–277. [Google Scholar] [CrossRef]
- Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef]
- Ren, Z.; Liang, H.; Galbo, P.M.; Dharmaratne, M.; Kulkarni, A.S.; Fard, A.T.; Aoun, M.L.; Martinez-Lopez, N.; Suyama, K.; Benard, O.; et al. Redox Signaling by Glutathione Peroxidase 2 Links Vascular Modulation to Metabolic Plasticity of Breast Cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2107266119. [Google Scholar] [CrossRef]
- Lee, E.; Choi, A.; Jun, Y.; Kim, N.; Yook, J.I.; Kim, S.Y.; Lee, S.; Kang, S.W. Glutathione Peroxidase-1 Regulates Adhesion and Metastasis of Triple-Negative Breast Cancer Cells via FAK Signaling. Redox Biol. 2020, 29, 101391. [Google Scholar] [CrossRef]
- Ahmadiankia, N.; Bagheri, M.; Fazli, M. Nutrient Deprivation Modulates the Metastatic Potential of Breast Cancer Cells. Rep. Biochem. Mol. Biol. 2019, 8, 139. [Google Scholar]
- Fernández-Sáiz, V.; Targosz, B.S.; Lemeer, S.; Eichner, R.; Langer, C.; Bullinger, L.; Reiter, C.; Slotta-Huspenina, J.; Schroeder, S.; Knorn, A.M.; et al. SCFFbxo9 and CK2 Direct the Cellular Response to Growth Factor Withdrawal via Tel2/Tti1 Degradation and Promote Survival in Multiple Myeloma. Nat. Cell Biol. 2012, 15, 72–81. [Google Scholar] [CrossRef]
- Park, J.W.; Jeong, J.; Bae, Y.S. Protein Kinase CK2 Is Upregulated by Calorie Restriction and Induces Autophagy. Mol. Cells 2022, 45, 112–121. [Google Scholar] [CrossRef]
- Townley, A.R.; Wheatley, S.P. Mitochondrial Survivin Reduces Oxidative Phosphorylation in Cancer Cells by Inhibiting Mitophagy. J. Cell Sci. 2020, 133, jcs247379. [Google Scholar] [CrossRef]
- Hauck, L.; Harms, C.; Rohne, J.; Gertz, K.; Dietz, R.; Endres, M.; Von Harsdorf, R. Protein Kinase CK2 Links Extracellular Growth Factor Signaling with the Control of P27(Kip1) Stability in the Heart. Nat. Med. 2008, 14, 315–324. [Google Scholar] [CrossRef]
- Sun, X.; Momen, A.; Wu, J.; Noyan, H.; Li, R.; Von Harsdorf, R.; Husain, M. P27 Protein Protects Metabolically Stressed Cardiomyocytes from Apoptosis by Promoting Autophagy. J. Biol. Chem. 2014, 289, 16924–16935. [Google Scholar] [CrossRef]
- Husain, K.; Williamson, T.T.; Nelson, N.; Ghansah, T. Protein Kinase 2 (CK2): A Potential Regulator of Immune Cell Development and Function in Cancer. Immunol. Med. 2021, 44, 159–174. [Google Scholar] [CrossRef]
- Aseervatham, J. Cytoskeletal Remodeling in Cancer. Biology 2020, 9, 385. [Google Scholar] [CrossRef]
- D’Amore, C.; Salizzato, V.; Borgo, C.; Cesaro, L.; Pinna, L.A.; Salvi, M. A Journey through the Cytoskeleton with Protein Kinase CK2. Curr. Protein Pept. Sci. 2019, 20, 547–562. [Google Scholar] [CrossRef]
- Azadi, S.; Tafazzoli Shadpour, M. The Microenvironment and Cytoskeletal Remodeling in Tumor Cell Invasion. Int. Rev. Cell Mol. Biol. 2020, 356, 257–289. [Google Scholar] [CrossRef]
- Daojing, W.; Jang, D.J. Protein Kinase CK2 Regulates Cytoskeletal Reorganization during Ionizing Radiation-Induced Senescence of Human Mesenchymal Stem Cells. Cancer Res. 2009, 69, 8200. [Google Scholar] [CrossRef]
- Dulyaninova, N.G.; House, R.P.; Betapudi, V.; Bresnick, A.R. Myosin-IIA Heavy-Chain Phosphorylation Regulates the Motility of MDA-MB-231 Carcinoma Cells. Mol. Biol. Cell 2007, 18, 3144–3155. [Google Scholar] [CrossRef]
- Canton, D.A.; Olsten, M.E.K.; Kim, K.; Doherty-Kirby, A.; Lajoie, G.; Cooper, J.A.; Litchfield, D.W. The Pleckstrin Homology Domain-Containing Protein CKIP-1 Is Involved in Regulation of Cell Morphology and the Actin Cytoskeleton and Interaction with Actin Capping Protein. Mol. Cell. Biol. 2005, 25, 3519–3534. [Google Scholar] [CrossRef]
- Loukil, A.; Barrington, C.; Goetz, S.C. A Complex of Distal Appendage-Associated Kinases Linked to Human Disease Regulates Ciliary Trafficking and Stability. Proc. Natl. Acad. Sci. USA 2021, 118, e2018740118. [Google Scholar] [CrossRef]
- Bowie, E.; Norris, R.; Anderson, K.V.; Goetz, S.C. Spinocerebellar Ataxia Type 11-Associated Alleles of Ttbk2 Dominantly Interfere with Ciliogenesis and Cilium Stability. PLoS Genet. 2018, 14, e1007844. [Google Scholar] [CrossRef]
- Liu, H.; Kiseleva, A.A.; Golemis, E.A. Ciliary Signaling in Cancer. Nat. Rev. Cancer 2018, 18, 511. [Google Scholar] [CrossRef]
- Cengiz, C.; Bulut, S.; Boyacioglu, A.S.; Kuzu, M.A. Nerve/Glial Antigen 2: A Novel Target for Anti-Tumor Therapy in Colorectal Cancer. Digestion 2017, 96, 60–66. [Google Scholar] [CrossRef]
- Yadavilli, S.; Hwang, E.I.; Packer, R.J.; Nazarian, J. The Role of NG2 Proteoglycan in Glioma. Transl. Oncol. 2016, 9, 57. [Google Scholar] [CrossRef]
- Lu, L.L.; Sun, J.; Lai, J.J.; Jiang, Y.; Bai, L.H.; Zhang, L. Da Neuron-Glial Antigen 2 Overexpression in Hepatocellular Carcinoma Predicts Poor Prognosis. World J. Gastroenterol. WJG 2015, 21, 6649. [Google Scholar] [CrossRef]
- Boewe, A.S.; Wemmert, S.; Kulas, P.; Schick, B.; Götz, C.; Wrublewsky, S.; Montenarh, M.; Menger, M.D.; Laschke, M.W.; Ampofo, E. Inhibition of CK2 Reduces NG2 Expression in Juvenile Angiofibroma. Biomedicines 2022, 10, 966. [Google Scholar] [CrossRef]
- Cattaruzza, S.; Nicolosi, P.A.; Braghetta, P.; Pazzaglia, L.; Benassi, M.S.; Picci, P.; Lacrima, K.; Zanocco, D.; Rizzo, E.; Stallcup, W.B.; et al. NG2/CSPG4-Collagen Type VI Interplays Putatively Involved in the Microenvironmental Control of Tumour Engraftment and Local Expansion. J. Mol. Cell Biol. 2013, 5, 176–193. [Google Scholar] [CrossRef]
- Watabe, M.; Nakaki, T. Protein Kinase CK2 Regulates the Formation and Clearance of Aggresomes in Response to Stress. J. Cell Sci. 2011, 124, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xiang, S.; Joo, H.Y.; Wang, L.; Williams, K.A.; Liu, W.; Hu, C.; Tong, D.; Haakenson, J.; Wang, C.; et al. HDAC6 Deacetylates and Ubiquitinates MSH2 to Maintain Proper Levels of MutSα. Mol. Cell 2014, 55, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, C.; Moses, N.; Haakenson, J.; Xiang, S.; Quan, D.; Fang, B.; Yang, Z.; Bai, W.; Bepler, G.; et al. HDAC6 Regulates DNA Damage Response via Deacetylating MLH1. J. Biol. Chem. 2019, 294, 5813–5826. [Google Scholar] [CrossRef] [PubMed]
- Banik, D.; Noonepalle, S.; Hadley, M.; Palmer, E.; Gracia-Hernandez, M.; Zevallos-Delgado, C.; Manhas, N.; Simonyan, H.; Young, C.N.; Popratiloff, A.; et al. HDAC6 Plays a Noncanonical Role in the Regulation of Antitumor Immune Responses, Dissemination, and Invasiveness of Breast Cancer. Cancer Res. 2020, 80, 3649–3662. [Google Scholar] [CrossRef]
- Borriello, L.; Karagiannis, G.S.; Duran, C.L.; Coste, A.; Oktay, M.H.; Entenberg, D.; Condeelis, J.S. The Role of the Tumor Microenvironment in Tumor Cell Intravasation and Dissemination. Eur. J. Cell Biol. 2020, 99, 151098. [Google Scholar] [CrossRef]
- Rathje, L.S.Z.; Nordgren, N.; Pettersson, T.; Rönnlund, D.; Widengren, J.; Aspenström, P.; Gad, A.K.B. Oncogenes Induce a Vimentin Filament Collapse Mediated by HDAC6 That Is Linked to Cell Stiffness. Proc. Natl. Acad. Sci. USA 2014, 111, 1515–1520. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, H.; Wang, J.; Liu, Y.; Luo, T.; Hua, H. Targeting Extracellular Matrix Stiffness and Mechanotransducers to Improve Cancer Therapy. J. Hematol. Oncol. 2022, 15, 34. [Google Scholar] [CrossRef]
- Lim, A.C.B.; Tiu, S.Y.; Li, Q.; Qi, R.Z. Direct Regulation of Microtubule Dynamics by Protein Kinase CK2. J. Biol. Chem. 2004, 279, 4433–4439. [Google Scholar] [CrossRef]
- Raab, M.; Discher, D.E. Matrix Rigidity Regulates the Microtubule Network Polarization in Migration. Cytoskeleton 2017, 74, 114. [Google Scholar] [CrossRef]
- You, E.; Jeong, J.; Lee, J.; Keum, S.; Hwang, Y.E.; Choi, J.-H.; Rhee, S. Casein Kinase 2 Promotes the TGF-β-Induced Activation of α-Tubulin Acetyltransferase 1 in Fibroblasts Cultured on a Soft Matrix. BMB Rep. 2022, 55, 192. [Google Scholar] [CrossRef]
- Boggs, A.E.; Vitolo, M.I.; Whipple, R.A.; Charpentier, M.S.; Goloubeva, O.G.; Ioffe, O.B.; Tuttle, K.C.; Slovic, J.; Lu, Y.; Mills, G.B.; et al. α-Tubulin Acetylation Elevated in Metastatic and Basal-like Breast Cancer Cells Promotes Microtentacle Formation, Adhesion, and Invasive Migration. Cancer Res. 2015, 75, 203–215. [Google Scholar] [CrossRef]
- You, E.; Ko, P.; Jeong, J.; Keum, S.; Kim, J.W.; Seo, Y.J.; Song, W.K.; Rhee, S. Dynein-Mediated Nuclear Translocation of Yes-Associated Protein through Microtubule Acetylation Controls Fibroblast Activation. Cell. Mol. Life Sci. CMLS 2020, 77, 4143–4161. [Google Scholar] [CrossRef]
- Andersen, A.P.; Moreira, J.M.A.; Pedersen, S.F. Interactions of Ion Transporters and Channels with Cancer Cell Metabolism and the Tumour Microenvironment. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130098. [Google Scholar] [CrossRef]
- Montenarh, M.; Götz, C. Protein Kinase CK2 and Ion Channels (Review). Biomed. Rep. 2020, 13, 55. [Google Scholar] [CrossRef]
- Crottès, D.; Jan, L.Y. The Multifaceted Role of TMEM16A in Cancer. Cell Calcium 2019, 82, 102050. [Google Scholar] [CrossRef]
- Pinto, M.C.; Schreiber, R.; Lerias, J.; Ousingsawat, J.; Duarte, A.; Amaral, M.; Kunzelmann, K. Regulation of TMEM16A by CK2 and Its Role in Cellular Proliferation. Cells 2020, 9, 1138. [Google Scholar] [CrossRef]
- Herrmann, D.; Straubinger, M.; Hashemolhosseini, S. Protein Kinase CK2 Interacts at the Neuromuscular Synapse with Rapsyn, Rac1, 14-3-3γ, and Dok-7 Proteins and Phosphorylates the Latter Two. J. Biol. Chem. 2015, 290, 22370–22384. [Google Scholar] [CrossRef]
- Simon, S.; Grabellus, F.; Ferrera, L.; Galietta, L.; Schwindenhammer, B.; Muḧlenberg, T.; Taeger, G.; Eilers, G.; Treckmann, J.; Breitenbuecher, F.; et al. DOG1 Regulates Growth and IGFBP5 in Gastrointestinal Stromal Tumors. Cancer Res. 2013, 73, 3661–3670. [Google Scholar] [CrossRef]
- Rodríguez, F.; Allende, C.C.; Allende, J.E. Protein Kinase Casein Kinase 2 Holoenzyme Produced Ectopically in Human Cells Can Be Exported to the External Side of the Cellular Membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 4718. [Google Scholar] [CrossRef]
- Rodriguez, F.A.; Contreras, C.; Bolanos-Garcia, V.; Allende, J.E. Protein Kinase CK2 as an Ectokinase: The Role of the Regulatory CK2beta Subunit. Proc. Natl. Acad. Sci. USA 2008, 105, 5693–5698. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 56, 15. [Google Scholar] [CrossRef]
- Lin, T.C.; Yang, C.H.; Cheng, L.H.; Chang, W.T.; Lin, Y.R.; Cheng, H.C. Fibronectin in Cancer: Friend or Foe. Cells 2020, 9, 27. [Google Scholar] [CrossRef]
- Yalak, G.; Vogel, V. Ectokinases as Novel Cancer Markers and Drug Targets in Cancer Therapy. Cancer Med. 2015, 4, 404. [Google Scholar] [CrossRef]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix Metalloproteinases (MMPs), the Main Extracellular Matrix (ECM) Enzymes in Collagen Degradation, as a Target for Anticancer Drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef]
- Filipiak, K.; Kubiński, K.; Hellman, U.; Ramos, A.; de Pascual-Teresa, B. Human Protein Kinase CK2 Phosphorylates Matrix Metalloproteinase 2 and Inhibits Its Activity. Chembiochem A Eur. J. Chem. Biol. 2014, 15, 1873–1876. [Google Scholar] [CrossRef]
- Dzobo, K. Integrins within the Tumor Microenvironment: Biological Functions, Importance for Molecular Targeting, and Cancer Therapeutics Innovation. OMICS A J. Integr. Biol. 2021, 25, 417–430. [Google Scholar] [CrossRef]
- Shishido, S.; Bönig, H.; Kim, Y.M. Role of Integrin Alpha4 in Drug Resistance of Leukemia. Front. Oncol. 2014, 4, 99. [Google Scholar] [CrossRef]
- Yao, E.S.; Zhang, H.; Chen, Y.Y.; Lee, B.; Chew, K.; Moore, D.; Park, C. Increased Beta1 Integrin Is Associated with Decreased Survival in Invasive Breast Cancer. Cancer Res. 2007, 67, 659–664. [Google Scholar] [CrossRef]
- Baiula, M.; Spampinato, S.; Gentilucci, L.; Tolomelli, A. Novel Ligands Targeting A4β1 Integrin: Therapeutic Applications and Perspectives. Front. Chem. 2019, 7, 489. [Google Scholar] [CrossRef]
- Burgos-Panadero, R.; Noguera, I.; Cañete, A.; Navarro, S.; Noguera, R. Vitronectin as a Molecular Player of the Tumor Microenvironment in Neuroblastoma. BMC Cancer 2019, 19, 479. [Google Scholar] [CrossRef]
- DiPersio, C.M.; Longmate, W. Beyond Adhesion: Emerging Roles for Integrins in Control of the Tumor Microenvironment. F1000Research 2017, 6, 1612. [Google Scholar] [CrossRef]
- López de Andrés, J.; Griñán-Lisón, C.; Jiménez, G.; Marchal, J.A. Cancer Stem Cell Secretome in the Tumor Microenvironment: A Key Point for an Effective Personalized Cancer Treatment. J. Hematol. Oncol. 2020, 13, 136. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A. Therapeutic Inhibition of Hedgehog-GLI Signaling in Cancer: Epithelial, Stromal, or Stem Cell Targets? Cancer Cell 2008, 14, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue Repair and Stem Cell Renewal in Carcinogenesis. Nature 2004, 432, 324–331. [Google Scholar] [CrossRef]
- Basu, S.; Cheriyamundath, S.; Ben-Ze’ev, A. Cell–Cell Adhesion: Linking Wnt/β-Catenin Signaling with Partial EMT and Stemness Traits in Tumorigenesis. F1000Research 2018, 7, 1488. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Zur Hausen, A.; et al. The EMT-Activator ZEB1 Promotes Tumorigenicity by Repressing Stemness-Inhibiting MicroRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Chung, D.W.D.; Frausto, R.F.; Ann, L.B.; Jang, M.S.; Aldave, A.J. Functional Impact of ZEB1 Mutations Associated With Posterior Polymorphous and Fuchs’ Endothelial Corneal Dystrophies. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6159. [Google Scholar] [CrossRef]
- Liu, Q.; Hodge, J.; Wang, J.; Wang, Y.; Wang, L.; Singh, U.; Li, Y.; Yao, Y.; Wang, D.; Ai, W.; et al. Emodin Reduces Breast Cancer Lung Metastasis by Suppressing Macrophage-Induced Breast Cancer Cell Epithelial-Mesenchymal Transition and Cancer Stem Cell Formation. Theranostics 2020, 10, 8365. [Google Scholar] [CrossRef]
- Zheng, X.; Yu, C.; Xu, M. Linking Tumor Microenvironment to Plasticity of Cancer Stem Cells: Mechanisms and Application in Cancer Therapy. Front. Oncol. 2021, 11, 2552. [Google Scholar] [CrossRef]
- Sato, K.; Padgaonkar, A.A.; Baker, S.J.; Cosenza, S.C.; Rechkoblit, O.; Subbaiah, D.R.C.V.; Domingo-Domenech, J.; Bartkowski, A.; Port, E.R.; Aggarwal, A.K.; et al. Simultaneous CK2/TNIK/DYRK1 Inhibition by 108600 Suppresses Triple Negative Breast Cancer Stem Cells and Chemotherapy-Resistant Disease. Nat. Commun. 2021, 12, 4671. [Google Scholar] [CrossRef]
- Agarwal, M.; Nitta, R.T.; Li, G. Casein Kinase 2: A Novel Player in Glioblastoma Therapy and Cancer Stem Cells. J. Mol. Genet. Med. Int. J. Biomed. Res. 2014, 8, 1000094. [Google Scholar] [CrossRef]
- Schupp, J.; Krebs, F.K.; Zimmer, N.; Trzeciak, E.; Schuppan, D.; Tuettenberg, A. Targeting Myeloid Cells in the Tumor Sustaining Microenvironment. Cell. Immunol. 2019, 343, 103713. [Google Scholar] [CrossRef]
- Cheng, P.; Kumar, V.; Liu, H.; Youn, J.I.; Fishman, M.; Sherman, S.; Gabrilovich, D. Effects of Notch Signaling on Regulation of Myeloid Cell Differentiation in Cancer. Cancer Res. 2014, 74, 141–152. [Google Scholar] [CrossRef]
- Hashimoto, A.; Gao, C.; Mastio, J.; Kossenkov, A.; Abrams, S.I.; Purandare, A.V.; Desilva, H.; Wee, S.; Hunt, J.; Jure-Kunkel, M.; et al. Inhibition of Casein Kinase 2 Disrupts Differentiation of Myeloid Cells in Cancer and Enhances the Efficacy of Immunotherapy in Mice. Cancer Res. 2018, 78, 5644. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Zhang, Y.; Dees, C.; Beyer, C.; Lin, N.Y.; Distler, A.; Zerr, P.; Palumbo, K.; Susok, L.; Kreuter, A.; Distler, O.; et al. Inhibition of Casein Kinase II Reduces TGFβ Induced Fibroblast Activation and Ameliorates Experimental Fibrosis. Ann. Rheum. Dis. 2015, 74, 936–943. [Google Scholar] [CrossRef]
- Skjerpen, C.S.; Nilsen, T.; Wesche, J.; Olsnes, S. Binding of FGF-1 Variants to Protein Kinase CK2 Correlates with Mitogenicity. EMBO J. 2002, 21, 4058–4069. [Google Scholar] [CrossRef]
- Bonnet, H.; Filhol, O.; Truchet, I.; Brethenou, P.; Cochet, C.; Amalric, F.; Bouche, G. Fibroblast Growth Factor-2 Binds to the Regulatory β Subunit of CK2 and Directly Stimulates CK2 Activity toward Nucleolin. J. Biol. Chem. 1996, 271, 24781–24787. [Google Scholar] [CrossRef]
- Xiao, S. The Role of Nucleolin Phosphorylation by CK2 in Regulating Cellular Fate under Normal and Stress Conditions. Ph.D. Thesis, City University of New York, New York, NY, USA, 2017. [Google Scholar]
- Carvalho, L.S.; Gonçalves, N.; Fonseca, N.A.; Moreira, J.N. Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesis. Pharmaceuticals 2021, 14, 60. [Google Scholar] [CrossRef]
- Gregório, A.C.; Lacerda, M.; Figueiredo, P.; Simões, S.; Dias, S.; Moreira, J.N. Meeting the Needs of Breast Cancer: A Nucleolin’s Perspective. Crit. Rev. Oncol./Hematol. 2018, 125, 89–101. [Google Scholar] [CrossRef]
- Li, Q.; Zong, Y.; Li, K.; Jie, X.; Hong, J.; Zhou, X.; Wu, B.; Li, Z.; Zhang, S.; Wu, G.; et al. Involvement of Endothelial CK2 in the Radiation Induced Perivascular Resistant Niche (PVRN) and the Induction of Radioresistance for Non-Small Cell Lung Cancer (NSCLC) Cells. Biol. Res. 2019, 52, 22. [Google Scholar] [CrossRef]
- Zhang, Y.; Tighe, S.; Zhu, Y.T. COX-2 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 87–104. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 Promotes Tumor Growth and Suppresses Tumor Immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Yefi, R.; Ponce, D.P.; Niechi, I.; Silva, E.; Cabello, P.; Rodriguez, D.A.; Marcelain, K.; Armisen, R.; Quest, A.F.G.; Tapia, J.C. Protein Kinase CK2 Promotes Cancer Cell Viability via Up-Regulation of Cyclooxygenase-2 Expression and Enhanced Prostaglandin E2 Production. J. Cell. Biochem. 2011, 112, 3167–3175. [Google Scholar] [CrossRef]
- Okwan-Duodu, D.; Landry, J.; Shen, X.Z.; Diaz, R. Angiotensin-Converting Enzyme and the Tumor Microenvironment: Mechanisms beyond Angiogenesis. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2013, 305, 205–215. [Google Scholar] [CrossRef]
- Kohlstedt, K.; Shoghi, F.; Müller-Esterl, W.; Busse, R.; Fleming, I. CK2 Phosphorylates the Angiotensin-Converting Enzyme and Regulates Its Retention in the Endothelial Cell Plasma Membrane. Circ. Res. 2002, 91, 749–756. [Google Scholar] [CrossRef]
- Kohlstedt, K.; Brandes, R.P.; Müller-Esterl, W.; Busse, R.; Fleming, I. Angiotensin-Converting Enzyme Is Involved in Outside-in Signaling in Endothelial Cells. Circ. Res. 2004, 94, 60–67. [Google Scholar] [CrossRef] [PubMed]
- De Alvarenga, E.C.; De Castro Fonseca, M.; Carvalho, C.C.; Florentino, R.M.; França, A.; Matias, E.; Guimarães, P.B.; Batista, C.; Freire, V.; Carmona, A.K.; et al. Angiotensin Converting Enzyme Regulates Cell Proliferation and Migration. PLoS ONE 2016, 11, e0165371. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The Hypoxic Tumour Microenvironment. Oncogenesis 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lin, D.; Taniguchi, C.M. Hypoxia Inducible Factor (HIF) in the Tumor Microenvironment: Friend or Foe? Sci. China. Life Sci. 2017, 60, 1114. [Google Scholar] [CrossRef]
- Li, L.Y.; Di Guan, Y.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 2520. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability—An Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, P.K. DNA Damage Repair: Historical Perspectives, Mechanistic Pathways and Clinical Translation for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef]
- Yi, C.; He, C. DNA Repair by Reversal of DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, 12575–12576. [Google Scholar] [CrossRef]
- Srivenugopal, K.S.; Mullapudi, S.R.; Shou, J.; Hazra, T.K.; Ali-Osman, F. Protein Phosphorylation Is a Regulatory Mechanism for O6-Alkylguanine-DNA Alkyltransferase in Human Brain Tumor Cells. Cancer Res. 2000, 60, 282–287. [Google Scholar]
- Yu, H.; Yang, X.; Tang, J.; Si, S.; Zhou, Z.; Lu, J.; Han, J.; Yuan, B.; Wu, Q.; Lu, Q.; et al. ALKBH5 Inhibited Cell Proliferation and Sensitized Bladder Cancer Cells to Cisplatin by M6A-CK2α-Mediated Glycolysis. Mol. Therapy. Nucleic Acids 2020, 23, 27–41. [Google Scholar] [CrossRef]
- Grundy, G.J.; Parsons, J.L. Base Excision Repair and Its Implications to Cancer Therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef]
- Parsons, J.L.; Dianova, I.I.; Finch, D.; Tait, P.S.; Ström, C.E.; Helleday, T.; Dianov, G.L. XRCC1 Phosphorylation by CK2 Is Required for Its Stability and Efficient DNA Repair. DNA Repair 2010, 9, 835–841. [Google Scholar] [CrossRef]
- Ström, C.E.; Mortusewicz, O.; Finch, D.; Parsons, J.L.; Lagerqvist, A.; Johansson, F.; Schultz, N.; Erixon, K.; Dianov, G.L.; Helleday, T. CK2 Phosphorylation of XRCC1 Facilitates Dissociation from DNA and Single-Strand Break Formation during Base Excision Repair. DNA Repair 2011, 10, 961–969. [Google Scholar] [CrossRef]
- Krohn, N.M.; Stemmer, C.; Fojan, P.; Grimm, R.; Grasser, K.D. Protein Kinase CK2 Phosphorylates the High Mobility Group Domain Protein SSRP1, Inducing the Recognition of UV-Damaged DNA. J. Biol. Chem. 2003, 278, 12710–12715. [Google Scholar] [CrossRef]
- Charles Richard, J.L.; Shukla, M.S.; Menoni, H.; Ouararhni, K.; Lone, I.N.; Roulland, Y.; Papin, C.; Ben Simon, E.; Kundu, T.; Hamiche, A.; et al. FACT Assists Base Excision Repair by Boosting the Remodeling Activity of RSC. PLoS Genet. 2016, 12, e1006221. [Google Scholar] [CrossRef]
- Li, Y.; Keller, D.M.; Scott, J.D.; Lu, H. CK2 Phosphorylates SSRP1 and Inhibits Its DNA-Binding Activity. J. Biol. Chem. 2005, 280, 11869–11875. [Google Scholar] [CrossRef]
- Keller, D.M.; Zeng, X.; Wang, Y.; Zhang, Q.H.; Kapoor, M.; Shu, H.; Goodman, R.; Lozano, G.; Zhao, Y.; Lu, H. A DNA Damage-Induced P53 Serine 392 Kinase Complex Contains CK2, HSpt16, and SSRP1. Mol. Cell 2001, 7, 283–292. [Google Scholar] [CrossRef]
- Keller, D.M.; Lu, H. P53 Serine 392 Phosphorylation Increases after UV through Induction of the Assembly of the CK2.HSPT16.SSRP1 Complex. J. Biol. Chem. 2002, 277, 50206–50213. [Google Scholar] [CrossRef]
- Castrogiovanni, C.; Waterschoot, B.; De Backer, O.; Dumont, P. Serine 392 Phosphorylation Modulates P53 Mitochondrial Translocation and Transcription-Independent Apoptosis. Cell Death Differ. 2017, 25, 190–203. [Google Scholar] [CrossRef]
- Araki, M.; Masutani, C.; Takemura, M.; Uchida, A.; Sugasawa, K.; Kondoh, J.; Ohkuma, Y.; Hanaoka, F. Centrosome Protein Centrin 2/Caltractin 1 Is Part of the Xeroderma Pigmentosum Group C Complex That Initiates Global Genome Nucleotide Excision Repair. J. Biol. Chem. 2001, 276, 18665–18672. [Google Scholar] [CrossRef]
- Shah, P.; Zhao, B.; Qiang, L.; He, Y.Y. Phosphorylation of Xeroderma Pigmentosum Group C Regulates Ultraviolet-Induced DNA Damage Repair. Nucleic Acids Res. 2018, 46, 5050–5060. [Google Scholar] [CrossRef]
- Grecu, D.; Assairi, L. CK2 Phosphorylation of Human Centrins 1 and 2 Regulates Their Binding to the DNA Repair Protein XPC, the Centrosomal Protein Sfi1 and the Phototransduction Protein Transducin β. FEBS Open Bio. 2014, 4, 407–419. [Google Scholar] [CrossRef]
- Kokic, G.; Chernev, A.; Tegunov, D.; Dienemann, C.; Urlaub, H.; Cramer, P. Structural Basis of TFIIH Activation for Nucleotide Excision Repair. Nat. Commun. 2019, 10, 2885. [Google Scholar] [CrossRef]
- Coin, F.; Auriol, J.; Tapias, A.; Clivio, P.; Vermeulen, W.; Egly, J.M. Phosphorylation of XPB Helicase Regulates TFIIH Nucleotide Excision Repair Activity. EMBO J. 2004, 23, 4835–4846. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Kartarius, S.; Schuster, N.; Montenarh, M. The Cyclin H/Cdk7/Mat1 Kinase Activity Is Regulated by CK2 Phosphorylation of Cyclin H. Oncogene 2002, 21, 5031–5037. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Lord, C.J.; Ashworth, A. Therapeutic Targeting of the DNA Mismatch Repair Pathway. Clin. Cancer Res. 2010, 16, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Tomicic, M.T.; Kaina, B. Phosphorylation of Mismatch Repair Proteins MSH2 and MSH6 Affecting MutSα Mismatch-Binding Activity. Nucleic Acids Res. 2002, 30, 1959. [Google Scholar] [CrossRef] [PubMed]
- Kaliyaperumal, S.; Patrick, S.M.; Williams, K.J. Phosphorylated HMSH6: DNA Mismatch versus DNA Damage Recognition. Mutat. Res. 2011, 706, 36. [Google Scholar] [CrossRef]
- Edelbrock, M.A.; Kaliyaperumal, S.; Williams, K.J. Structural, Molecular and Cellular Functions of MSH2 and MSH6 during DNA Mismatch Repair, Damage Signaling and Other Noncanonical Activities. Mutat. Res. 2013, 743, 53–66. [Google Scholar] [CrossRef]
- Wu, Q.; Huang, Y.; Gu, L.; Chang, Z.; Li, G.M. OTUB1 Stabilizes Mismatch Repair Protein MSH2 by Blocking Ubiquitination. J. Biol. Chem. 2021, 296, 100466. [Google Scholar] [CrossRef]
- Padilla-Benavides, T.; Nasipak, B.T.; Paskavitz, A.L.; Haokip, D.T.; Schnabl, J.M.; Nickerson, J.A.; Imbalzano, A.N. Casein Kinase 2-Mediated Phosphorylation of Brahma-Related Gene 1 Controls Myoblast Proliferation and Contributes to SWI/SNF Complex Composition. J. Biol. Chem. 2017, 292, 18592–18607. [Google Scholar] [CrossRef]
- Padilla-Benavides, T.; Haokip, D.T.; Yoon, Y.; Reyes-Gutierrez, P.; Rivera-Pérez, J.A.; Imbalzano, A.N. CK2-Dependent Phosphorylation of the Brg1 Chromatin Remodeling Enzyme Occurs during Mitosis. Int. J. Mol. Sci. 2020, 21, 923. [Google Scholar] [CrossRef]
- Nargund, A.M.; Xu, C.; Mandoli, A.; Okabe, A.; Chen, G.B.; Huang, K.K.; Sheng, T.; Yao, X.; Teo, J.M.N.; Sundar, R.; et al. Chromatin Rewiring by Mismatch Repair Protein MSH2 Alters Cell Adhesion Pathways and Sensitivity to BET Inhibition in Gastric Cancer. Cancer Res. 2022, 82, 2538–2551. [Google Scholar] [CrossRef]
- Weßbecher, I.M.; Hinrichsen, I.; Funke, S.; Oellerich, T.; Plotz, G.; Zeuzem, S.; Grus, F.H.; Biondi, R.M.; Brieger, A. DNA Mismatch Repair Activity of MutLα Is Regulated by CK2-Dependent Phosphorylation of MLH1 (S477). Mol. Carcinog. 2018, 57, 1723–1734. [Google Scholar] [CrossRef]
- Ulreich, K.; Firnau, M.B.; Tagscherer, N.; Beyer, S.; Ackermann, A.; Brieger, A.; Plotz, G. High Expression of Casein Kinase 2 Alpha Is Responsible for Enhanced Phosphorylation of DNA Mismatch Repair Protein MLH1 and Increased Tumor Mutation Rates in Colorectal Cancer. Cancers 2022, 14, 1553. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch Repair Deficiency/Microsatellite Instability-High as a Predictor for Anti-PD-1/PD-L1 Immunotherapy Efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Mills, A.M.; Dill, E.A.; Moskaluk, C.A.; Dziegielewski, J.; Bullock, T.N.; Dillon, P.M. The Relationship Between Mismatch Repair Deficiency and PD-L1 Expression in Breast Carcinoma. Am. J. Surg. Pathol. 2018, 42, 183–191. [Google Scholar] [CrossRef]
- Gong, J.; Wang, C.; Lee, P.P.; Chu, P.; Fakih, M. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring a POLE Mutation. J. Natl. Compr. Cancer Netw. JNCCN 2017, 15, 142–147. [Google Scholar] [CrossRef]
- Fabrizio, D.A.; George, T.J.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond Microsatellite Testing: Assessment of Tumor Mutational Burden Identifies Subsets of Colorectal Cancer Who May Respond to Immune Checkpoint Inhibition. J. Gastrointest. Oncol. 2018, 9, 610. [Google Scholar] [CrossRef]
- Xia, L.; Qin, K.; Wang, X.R.; Wang, X.L.; Zhou, A.W.; Chen, G.Q.; Lu, Y. Pyruvate Kinase M2 Phosphorylates H2AX and Promotes Genomic Instability in Human Tumor Cells. Oncotarget 2017, 8, 109120. [Google Scholar] [CrossRef]
- Liu, S.; Hua, Y.; Wang, J.; Li, L.; Yuan, J.; Zhang, B.; Wang, Z.; Ji, J.; Kong, D. RNA Polymerase III Is Required for the Repair of DNA Double-Strand Breaks by Homologous Recombination. Cell 2021, 184, 1314–1329.e10. [Google Scholar] [CrossRef]
- Ghavidel, A.; Schultz, M.C. TATA Binding Protein-Associated CK2 Transduces DNA Damage Signals to the RNA Polymerase III Transcriptional Machinery. Cell 2001, 106, 575–584. [Google Scholar] [CrossRef]
- Johnston, I.M.; Allison, S.J.; Morton, J.P.; Schramm, L.; Scott, P.H.; White, R.J. CK2 Forms a Stable Complex with TFIIIB and Activates RNA Polymerase III Transcription in Human Cells. Mol. Cell. Biol. 2002, 22, 3757. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495. [Google Scholar] [CrossRef]
- Koch, C.A.; Agyei, R.; Galicia, S.; Metalnikov, P.; O’Donnell, P.; Starostine, A.; Weinfeld, M.; Durocher, D. Xrcc4 Physically Links DNA End Processing by Polynucleotide Kinase to DNA Ligation by DNA Ligase IV. EMBO J. 2004, 23, 3874–3885. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 Complex Alterations and DNA Damage Response: Implications for Cancer Treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef]
- Von Morgen, P.V.; Burdova, K.; Flower, T.G.; O’Reilly, N.J.; Boulton, S.J.; Smerdon, S.J.; MacUrek, L.; Hoøejší, Z. MRE11 Stability Is Regulated by CK2-Dependent Interaction with R2TP Complex. Oncogene 2017, 36, 4943–4950. [Google Scholar] [CrossRef]
- Wu, L.; Luo, K.; Lou, Z.; Chen, J. MDC1 Regulates Intra-S-Phase Checkpoint by Targeting NBS1 to DNA Double-Strand Breaks. Proc. Natl. Acad. Sci. USA 2008, 105, 11200–11205. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sarkar, B.; Cholia, R.P.; Gautam, N.; Dhiman, M.; Mantha, A.K. APE1/Ref-1 as an Emerging Therapeutic Target for Various Human Diseases: Phytochemical Modulation of Its Functions. Exp. Mol. Med. 2014, 46, e106. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, A.; Kelley, M.R.; Deutsch, W.A. The DNA Repair Activity of Human Redox/Repair Protein APE/Ref-1 Is Inactivated by Phosphorylation. Cancer Res. 1997, 57, 5457–5459. [Google Scholar]
- Fritz, G.; Kaina, B. Phosphorylation of the DNA Repair Protein APE/REF-1 by CKII Affects Redox Regulation of AP-1. Oncogene 1999, 18, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Palma, A.; Pugliese, G.M.; Murfuni, I.; Marabitti, V.; Malacaria, E.; Rinalducci, S.; Minoprio, A.; Sanchez, M.; Mazzei, F.; Zolla, L.; et al. Phosphorylation by CK2 Regulates MUS81/EME1 in Mitosis and after Replication Stress. Nucleic Acids Res. 2018, 46, 5109–5124. [Google Scholar] [CrossRef]
- Ayoub, N.; Jeyasekharan, A.D.; Bernal, J.A.; Venkitaraman, A.R. HP1-Beta Mobilization Promotes Chromatin Changes That Initiate the DNA Damage Response. Nature 2008, 453, 682–686. [Google Scholar] [CrossRef]
- Onn, L.; Portillo, M.; Ilic, S.; Cleitman, G.; Stein, D.; Kaluski, S.; Shirat, I.; Slobodnik, Z.; Einav, M.; Erdel, F.; et al. SIRT6 Is a DNA Double-Strand Break Sensor. eLife 2020, 9, e51636. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Firsanov, D.; Zhang, Z.; Cheng, Y.; Luo, L.; Tombline, G.; Tan, R.; Simon, M.; Henderson, S.; Steffan, J.; et al. SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 2019, 177, 622–638.e22. [Google Scholar] [CrossRef] [PubMed]
- Kolinjivadi, A.M.; Chong, S.T.; Ngeow, J. Molecular Connections between Circadian Rhythm and Genome Maintenance Pathways. Endocr.-Relat. Cancer 2021, 28, R55–R66. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Cao, Z.; Zhang, J.; Tang, M.; Tian, Y.; Li, Y.; Lu, X.; Chen, Y.; Wang, H.; Wei, F.Z.; et al. SIRT6 Coordinates with CHD4 to Promote Chromatin Relaxation and DNA Repair. Nucleic Acids Res. 2020, 48, 2982–3000. [Google Scholar] [CrossRef]
- Shadan, F.F. Circadian Tempo: A Paradigm for Genome Stability? Med. Hypotheses 2007, 68, 883–891. [Google Scholar] [CrossRef]
- Fu, L.; Kettner, N.M. The Circadian Clock in Cancer Development and Therapy. Prog. Mol. Biol. Transl. Sci. 2013, 119, 221–282. [Google Scholar] [CrossRef]
- Okamoto-Uchida, Y.; Izawa, J.; Hirayama, J. A Molecular Link between the Circadian Clock, DNA Damage Responses, and Oncogene Activation. Oncog. Carcinog. 2018. [Google Scholar] [CrossRef]
- Tamaru, T.; Hirayama, J.; Isojima, Y.; Nagai, K.; Norioka, S.; Takamatsu, K.; Sassone-Corsi, P. Circadian Rhythmic Kinase CK2α Phosphorylates BMAL1 to Regulate the Mammalian Clock. Nat. Preced. 2008, 3, 1–17. [Google Scholar] [CrossRef]
- Tamaru, T.; Hattori, M.; Honda, K.; Nakahata, Y.; Sassone-Corsi, P.; van der Horst, G.T.J.; Ozawa, T.; Takamatsu, K. CRY Drives Cyclic CK2-Mediated BMAL1 Phosphorylation to Control the Mammalian Circadian Clock. PLoS Biol. 2015, 13, e1002293. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Akashi, M.; Matsuda, M.; Goto, K.; Miyata, Y.; Node, K.; Nishida, E. Involvement of the Protein Kinase CK2 in the Regulation of Mammalian Circadian Rhythms. Sci. Signal. 2009, 2, ra26. [Google Scholar] [CrossRef]
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell 2008, 134, 317–328. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef]
- O’Hagan, H.M. Chromatin Modifications during Repair of Environmental Exposure-Induced DNA Damage: A Potential Mechanism for Stable Epigenetic Alterations. Environ. Mol. Mutagenesis 2014, 55, 278–291. [Google Scholar] [CrossRef]
- Pyerin, W.; Barz, T.; Ackermann, K. Protein Kinase CK2 in Gene Control at Cell Cycle Entry. Mol. Cell. Biochem. 2005, 274, 189–200. [Google Scholar] [CrossRef]
- Feng, H.; Lu, J.; Song, X.; Thongkum, A.; Zhang, F.; Lou, L.; Reizes, O.; Almasan, A.; Gong, Z. CK2 Kinase-Mediated PHF8 Phosphorylation Controls TopBP1 Stability to Regulate DNA Replication. Nucleic Acids Res. 2020, 48, 10940–10952. [Google Scholar] [CrossRef]
- Ahuja, A.K.; Jodkowska, K.; Teloni, F.; Bizard, A.H.; Zellweger, R.; Herrador, R.; Ortega, S.; Hickson, I.D.; Altmeyer, M.; Mendez, J.; et al. A Short G1 Phase Imposes Constitutive Replication Stress and Fork Remodelling in Mouse Embryonic Stem Cells. Nat. Commun. 2016, 7, 10660. [Google Scholar] [CrossRef]
- Marchal, C.; Sima, J.; Gilbert, D.M. Control of DNA Replication Timing in the 3D Genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 721–737. [Google Scholar] [CrossRef]
- Allen, C.; Her, S.; Jaffray, D.A. Radiotherapy for Cancer: Present and Future. Adv. Drug Deliv. Rev. 2017, 109, 1–2. [Google Scholar] [CrossRef]
- Rajora, A.K.; Ravishankar, D.; Zhang, H.; Rosenholm, J.M. Recent Advances and Impact of Chemotherapeutic and Antiangiogenic Nanoformulations for Combination Cancer Therapy. Pharmaceutics 2020, 12, 592. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA Repair Pathways as Targets for Cancer Therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Xu, G.; Gou, S. Novel CK2-Specific Pt(II) Compound Reverses Cisplatin-Induced Resistance by Inhibiting Cancer Cell Stemness and Suppressing DNA Damage Repair in Non-Small Cell Lung Cancer Treatments. J. Med. Chem. 2021, 64, 4163–4178. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, P.; Zou, L.; Zhang, J.; Chen, J.; Yang, C.; He, G.; Liu, B.; Liu, J.; Chiang, C.M.; et al. Discovery of Novel Dual-Target Inhibitor of Bromodomain-Containing Protein 4/Casein Kinase 2 Inducing Apoptosis and Autophagy-Associated Cell Death for Triple-Negative Breast Cancer Therapy. J. Med. Chem. 2021, 64, 18025–18053. [Google Scholar] [CrossRef]
- Giacosa, S.; Pillet, C.; Séraudie, I.; Guyon, L.; Wallez, Y.; Roelants, C.; Battail, C.; Evrard, B.; Chalmel, F.; Barette, C.; et al. Cooperative Blockade of CK2 and ATM Kinases Drives Apoptosis in VHL-Deficient Renal Carcinoma Cells through ROS Overproduction. Cancers 2021, 13, 576. [Google Scholar] [CrossRef]
- Kildey, K.; Gandhi, N.S.; Sahin, K.B.; Shah, E.T.; Boittier, E.; Duijf, P.H.G.; Molloy, C.; Burgess, J.T.; Beard, S.; Bolderson, E.; et al. Elevating CDCA3 Levels in Non-Small Cell Lung Cancer Enhances Sensitivity to Platinum-Based Chemotherapy. Commun. Biol. 2021, 4, 638. [Google Scholar] [CrossRef]
- Nitta, R.T.; Bolin, S.; Luo, E.; Solow-Codero, D.E.; Samghabadi, P.; Purzner, T.; Aujla, P.S.; Nwagbo, G.; Cho, Y.J.; Li, G. Casein Kinase 2 Inhibition Sensitizes Medulloblastoma to Temozolomide. Oncogene 2019, 38, 6867. [Google Scholar] [CrossRef]
- Yu, W.-H. Development of Rational Combination Therapy with Parp Inhibitors and Kinase Inhibitors in Tnbc; The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access): Austin, TX, USA, 2016. [Google Scholar]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Shihab, I.; Khalil, B.A.; Elemam, N.M.; Hachim, I.Y.; Hachim, M.Y.; Hamoudi, R.A.; Maghazachi, A.A. Understanding the Role of Innate Immune Cells and Identifying Genes in Breast Cancer Microenvironment. Cancers 2020, 12, 2226. [Google Scholar] [CrossRef]
- Maiorino, L.; Daßler-Plenker, J.; Sun, L.; Egeblad, M. Innate Immunity and Cancer Pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 425–457. [Google Scholar] [CrossRef]
- Larson, S.R.; Bortell, N.; Illies, A.; Crisler, W.J.; Matsuda, J.L.; Lenz, L.L. Myeloid Cell CK2 Regulates Inflammation and Resistance to Bacterial Infection. Front. Immunol. 2020, 11, 3166. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 in the Pathogenesis and Treatment of Inflammatory Diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Liang, M.D.; Zhang, Y.; Mcdevit, D.; Marecki, S.; Nikolajczyk, B.S. The Interleukin-1 Gene Is Transcribed from a Poised Promoter Architecture in Monocytes. J. Biol. Chem. 2006, 281, 9227–9237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Saccani, S.; Shin, H.; Nikolajczyk, B.S. Dynamic Protein Associations Define Two Phases of IL-1β Transcriptional Activation. J. Immunol. 2008, 181, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.H.; Park, H.J.; Byun, H.E.; Park, Y.M.; Kim, T.W.; Kim, B.O.; Um, S.H.; Pyo, S. Diosgenin Inhibits Macrophage-Derived Inflammatory Mediators through Downregulation of CK2, JNK, NF-ΚB and AP-1 Activation. Int. Immunopharmacol. 2010, 10, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- De Bourayne, M.; Gallais, Y.; El Ali, Z.; Rousseau, P.; Héì Ene Damiens, M.; Cochet, C.; Filhol, O.; Chollet-Martin, S.; Pallardy, M.; Kerdine-Römer, S. Protein Kinase CK2 Controls T Cell Polarization through Dendritic Cells Activation in Response to Contact Sensitizers Protein Kinase CK2 Controls T-Cell Polarization through Dendritic Cell Activation in Response to Contact Sensitizers. J. Leukoc. Biol. 2017, 101, 703–715. [Google Scholar] [CrossRef]
- Reverendo, M.; Argüello, R.J.; Polte, C.; Valečka, J.; Camosseto, V.; Auphan-Anezin, N.; Ignatova, Z.; Gatti, E.; Pierre, P. Polymerase III Transcription Is Necessary for T Cell Priming by Dendritic Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 22721–22729. [Google Scholar] [CrossRef]
- Voisinne, G.; Gonzalez de Peredo, A.; Roncagalli, R. CD5, an Undercover Regulator of TCR Signaling. Front. Immunol. 2018, 9, 2900. [Google Scholar] [CrossRef]
- Sestero, C.M.; Mcguire, D.J.; De Sarno, P.; Brantley, E.C.; Soldevila, G.; Axtell, R.C.; Raman, C. CD5-Dependent CK2 Activation Pathway Regulates Threshold for T-Cell Anergy 1. J. Immunol. 2012, 189, 2918–2930. [Google Scholar] [CrossRef]
- Axtell, R.C.; Xu, L.; Barnum, S.R.; Raman, C. CD5-CK2 Binding/Activation Deficient Mice Are Resistant to EAE: Protection Is Associated with Diminished Populations of IL-17 Expressing T-Cells in the CNS. J. Immunol. 2006, 177, 8542–8549. [Google Scholar] [CrossRef]
- Ulges, A.; Klein, M.; Reuter, S.; Gerlitzki, B.; Hoffmann, M.; Grebe, N.; Staudt, V.; Stergiou, N.; Bohn, T.; Brühl, T.J.; et al. Protein Kinase CK2 Enables Regulatory T Cells to Suppress Excessive TH2 Responses in Vivo. Nat. Immunol. 2015, 16, 267–275. [Google Scholar] [CrossRef]
- Gibson, S.A.; Yang, W.; Yan, Z.; Liu, Y.; Rowse, A.L.; Weinmann, A.S.; Qin, H.; Benveniste, E.N. Protein Kinase CK2 Controls the Fate Between Th17 Cell and Regulatory T Cell Differentiation CK2 Regulates the Th17/Treg Axis. J. Immunol. 2017, 198, 4244–4254. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int. J. Mol. Sci. 2020, 21, 8011. [Google Scholar] [CrossRef]
- Ulges, A.; Witsch, E.J.; Pramanik, G.; Klein, M.; Birkner, K.; Bühler, U.; Wasser, B.; Luessi, F.; Stergiou, N.; Dietzen, S.; et al. Protein Kinase CK2 Governs the Molecular Decision between Encephalitogenic T H 17 Cell and T Reg Cell Development. Proc. Natl. Acad. Sci. USA 2016, 113, 10145–10150. [Google Scholar] [CrossRef]
- Low, D.; Mino-Kenudson, M.; Mizoguchi, E. Recent Advances in Understanding Colitis-Associated Tumorigenesis. Inflamm. Bowel. Dis. 2014, 20, 2115–2123. [Google Scholar] [CrossRef]
- Yang, W.; Gibson, S.A.; Yan, Z.; Wei, H.; Tao, J.; Bingdong, S.; Qin, H.; Benveniste, E.N. Protein Kinase 2 (CK2) Controls CD4 + T-Cell Effector Function in the Pathogenesis of Colitis. Mucosal Immunol. 2020, 13, 788–798. [Google Scholar] [CrossRef]
- Wei, H.; Yang, W.; Hong, H.; Yan, Z.; Qin, H.; Benveniste, E.N. Protein Kinase CK2 Regulates B Cell Development and Differentiation. J. Immunol. 2021, 207, 799–808. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, K.; Lee, K.H.; Kim, S.J.; Kim, J. Inhibition of Casein Kinase 2 Enhances the Death Ligand- and Natural Kiler Cell-Induced Hepatocellular Carcinoma Cell Death. Clin. Exp. Immunol. 2008, 152, 336–344. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-Educating” Tumor-Associated Macrophages by Targeting NF-KappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt Signaling Arrests Effector T Cell Differentiation and Generates CD8 + Memory Stem Cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef]
- Nelson, N.; Xiang, S.; Zhang, X.; Gilvary, D.; Djeu, J.; Husain, K.; Malafa, M.; Vohra, N.; Pilon-Thomas, S.; Ghansah, T. Murine Pancreatic Adenocarcinoma Reduces Ikaros Expression and Disrupts T Cell Homeostasis. PLoS ONE 2015, 10, e0115546. [Google Scholar] [CrossRef]
- Nelson, N.; Szekeres, K.; Iclozan, C.; Rivera, I.O.; Mcgill, A.; Johnson, G.; Nwogu, O.; Ghansah, T. Apigenin: Selective CK2 Inhibitor Increases Ikaros Expression and Improves T Cell Homeostasis and Function in Murine Pancreatic Cancer. PLoS ONE 2017, 12, e0170197. [Google Scholar] [CrossRef]
- Borgo, C.; D’Amore, C.; Cesaro, L.; Itami, K.; Hirota, T.; Salvi, M.; Pinna, L.A. A N-Terminally Deleted Form of the CK2α’ Catalytic Subunit Is Sufficient to Support Cell Viability. Biochem. Biophys. Res. Commun. 2020, 531, 409–415. [Google Scholar] [CrossRef]
- Cozza, G.; Meggio, F.; Moro, S. The Dark Side of Protein Kinase CK2 Inhibition. Curr. Med. Chem. 2011, 18, 2867–2884. [Google Scholar] [CrossRef]
- Qiao, Y.; Chen, T.; Yang, H.; Chen, Y.; Lin, H.; Qu, W.; Feng, F.; Liu, W.; Guo, Q.; Liu, Z.; et al. Small Molecule Modulators Targeting Protein Kinase CK1 and CK2. Eur. J. Med. Chem. 2019, 181, 111581. [Google Scholar] [CrossRef]
- Borgo, C.; Ruzzene, M. Chapter Two—Protein Kinase CK2 Inhibition as a Pharmacological Strategy. In Advances in Protein Chemistry and Structural Biology; Protein Kinases in Drug, Discovery; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2021; Volume 124, pp. 23–46. [Google Scholar]
- Salvi, M.; Borgo, C.; Pinna, L.A.; Ruzzene, M. Targeting CK2 in Cancer: A Valuable Strategy or a Waste of Time? Cell Death Discov. 2021, 7, 325. [Google Scholar] [CrossRef]
- Licciardello, M.P.; Workman, P. A New Chemical Probe Challenges the Broad Cancer Essentiality of CK2. Trends Pharmacol. Sci. 2021, 42, 313–315. [Google Scholar] [CrossRef]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-Chlorophenylamino)Benzo[c][2,6]Naphthyridine-8-Carboxylic Acid (CX-4945), the First Clinical Stage Inhibitor of Protein Kinase CK2 for the Treatment of Cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef]
- Wells, C.I.; Drewry, D.H.; Pickett, J.E.; Tjaden, A.; Krämer, A.; Müller, S.; Gyenis, L.; Menyhart, D.; Litchfield, D.W.; Knapp, S.; et al. Development of a Potent and Selective Chemical Probe for the Pleiotropic Kinase CK2. Cell Chem. Biol. 2021, 28, 546–558.e10. [Google Scholar] [CrossRef]
- Perea, S.E.; Baladrón, I.; Valenzuela, C.; Perera, Y. CIGB-300: A Peptide-Based Drug That Impairs the Protein Kinase CK2-Mediated Phosphorylation. Semin. Oncol. 2018, 45, 58–67. [Google Scholar] [CrossRef]
- D’Amore, C.; Borgo, C.; Sarno, S.; Salvi, M. Role of CK2 Inhibitor CX-4945 in Anti-Cancer Combination Therapy—Potential Clinical Relevance. Cell. Oncol. 2020, 43, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Firnau, M.-B.; Brieger, A. CK2 and the Hallmarks of Cancer. Biomedicines 2022, 10, 1987. https://doi.org/10.3390/biomedicines10081987
Firnau M-B, Brieger A. CK2 and the Hallmarks of Cancer. Biomedicines. 2022; 10(8):1987. https://doi.org/10.3390/biomedicines10081987
Chicago/Turabian StyleFirnau, May-Britt, and Angela Brieger. 2022. "CK2 and the Hallmarks of Cancer" Biomedicines 10, no. 8: 1987. https://doi.org/10.3390/biomedicines10081987
APA StyleFirnau, M. -B., & Brieger, A. (2022). CK2 and the Hallmarks of Cancer. Biomedicines, 10(8), 1987. https://doi.org/10.3390/biomedicines10081987