

Transcriptional Profiling of a Patient-Matched Cohort of Glioblastoma (IDH-Wildtype) for Therapeutic Target and Repurposing Drug Identification
 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Molecular Profiling of GBM IDH-Wildtype Tumors
2.2. RNA Extraction, Microarray Profiling, and Data Quality Control
2.3. Provisional Comparisons of Initial and Recurrent GBM
2.4. Differential Gene Expression Analysis between Initial and Recurrent GBM
2.5. Functional Enrichment Analyses
2.5.1. Gene Ontology Enrichment Analysis of the DEGs
2.5.2. Canonical Pathway Analysis
2.6. Upstream Regulator Effects
2.7. Gene Expression Connectivity Mapping
3. Results
3.1. Samples, Data Quality Control, and Provisional Comparisons of Initial and recGBM
3.2. Differential Gene Expression Analysis between Initial and recGBM
3.3. Functional Enrichment Analyses of the DEGs
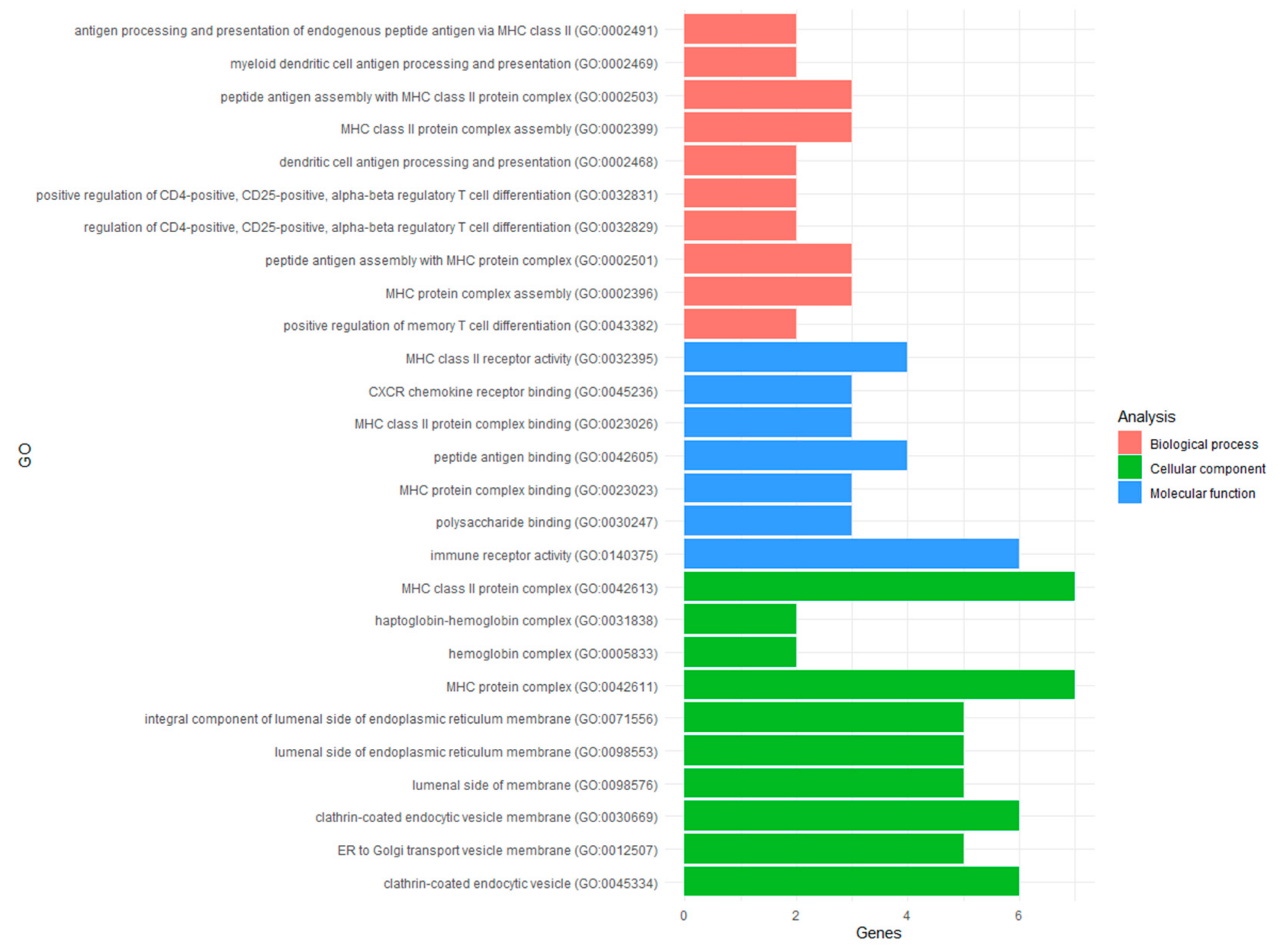
3.3.1. Gene Ontology Analysis
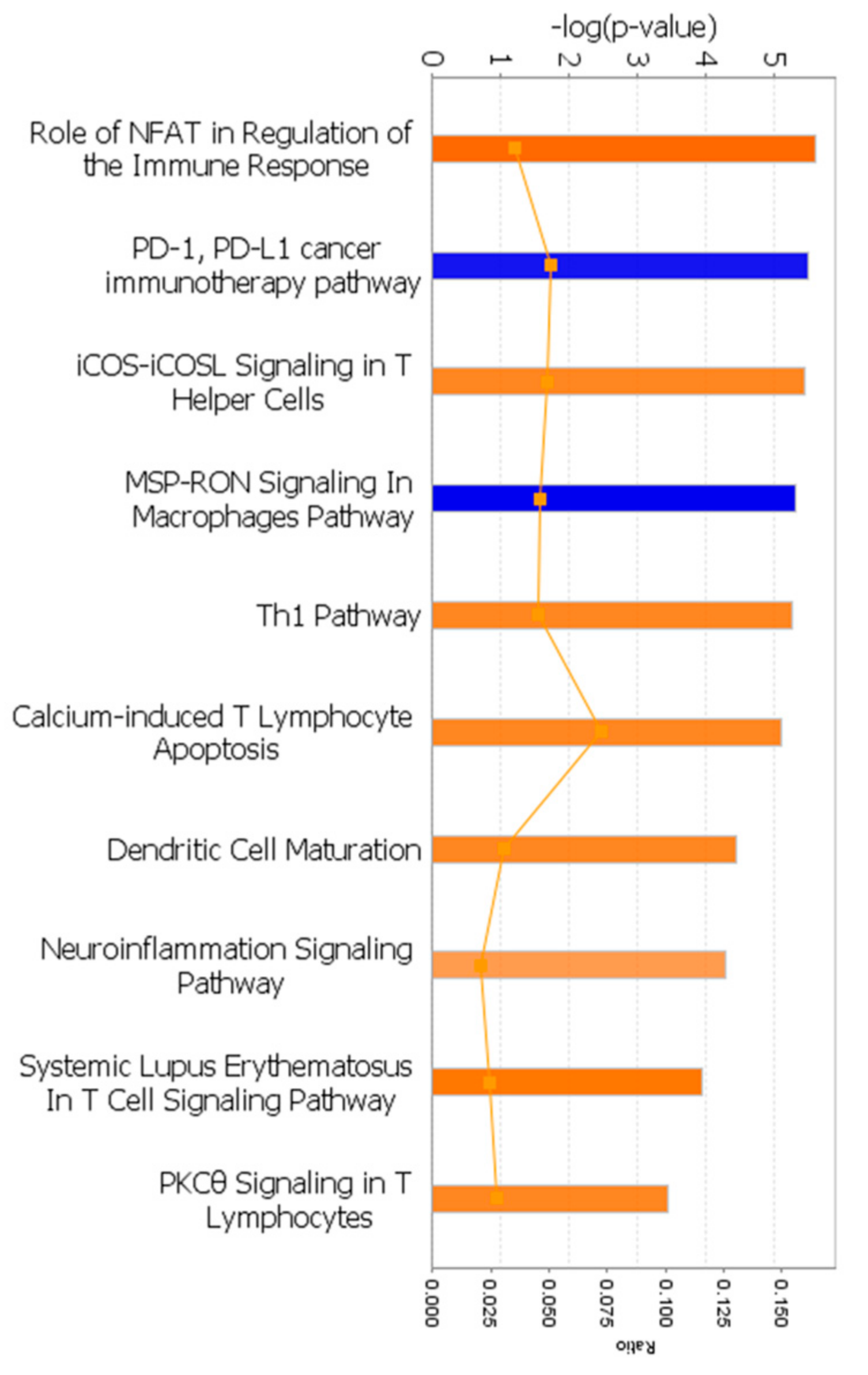
3.3.2. Canonical Pathway Analysis
3.4. Upstream Regulator Effects
3.5. Gene Expression Connectivity Mapping
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. Neuro-Oncol. 2021, 23 (Suppl. S3), iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma stem cells: Signaling, microenvironment, and therapy. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Fabbri, P.V.; Morandi, L.; de Biase, D.; di Oto, E.; Tallini, G.; Sturiale, C.; Franceschi, E.; Frezza, G.P.; Foschini, M.P. Pathological spectrum in recurrences of glioblastoma multiforme. Pathologica 2015, 107, 1–8. [Google Scholar]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O. Nonsurgical treatment of recurrent glioblastoma. Curr. Oncol. 2015, 22, 273–281. [Google Scholar] [CrossRef]
- Kwon, S.M.; Kang, S.H.; Park, C.K.; Jung, S.; Park, E.S.; Lee, J.S.; Kim, S.H.; Woo, H.G. Recurrent glioblastomas reveal molecular subtypes associated with mechanistic implications of drug-resistance. PLoS ONE 2015, 10, e0140528. [Google Scholar] [CrossRef]
- Kim, E.L.; Sorokin, M.; Kantelhardt, S.R.; Kalasauskas, D.; Sprang, B.; Fauss, J.; Ringel, F.; Garazha, A.; Albert, E.; Gaifullin, N.; et al. Intratumoral heterogeneity and longitudinal changes in gene expression predict differential drug sensitivity in newly diagnosed and recurrent glioblastoma. Cancers 2020, 12, 520. [Google Scholar] [CrossRef]
- Varn, F.S.; Johnson, K.C.; Martinek, J.; Huse, J.T.; Nasrallah, M.P.; Wesseling, P.; Cooper, L.A.; Malta, T.M.; Wade, T.E.; Sabedot, T.S.; et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 2022, 185, 2184–2199. [Google Scholar] [CrossRef]
- Alves, A.L.V.; Gomes, I.N.; Carloni, A.C.; Rosa, M.N.; da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of glioblastoma stem cells in cancer therapeutic resistance: A perspective on antineoplastic agents from natural sources and chemical derivatives. Stem Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- O’Reilly, P.G.; Wen, Q.; Bankhead, P.; Dunne, P.D.; McArt, D.G.; McPherson, S.; Hamilton, P.W.; Mills, K.I.; Zhang, S.D. QUADrATiC: Scalable gene expression connectivity mapping for repurposing FDA-approved therapeutics. BMC Bioinform. 2016, 17, 198. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A comprehensive resource with functional genomic data from Chinese glioma patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; DeCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. World Health Organization Classification of Tumours of the Central Nervous System, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2021. [Google Scholar]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Fears, C.Y.; Gladson, C.L.; Woods, A. Syndecan-2 is expressed in the microvasculature of gliomas and regulates angiogenic processes in microvascular endothelial cells. J. Biol. Chem. 2006, 281, 14533–14536. [Google Scholar] [CrossRef] [PubMed]
- Tabouret, E.; Tchoghandjian, A.; Denicolai, E.; Delfino, C.; Metellus, P.; Graillon, T.; Boucard, C.; Nanni, I.; Padovani, L.; Ouafik, L.H.; et al. Recurrence of glioblastoma after radio-chemotherapy is associated with an angiogenic switch to the CXCL12-CXCR4 pathway. Oncotarget 2015, 6, 11664. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and bevacizumab in progressive glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Diao, J.; Xia, T.; Zhao, H.; Liu, J.; Li, B.; Zhang, Z. Overexpression of HLA-DR is associated with prognosis of glioma patients. Int. J. Clin. Exp. Pathol. 2015, 8, 5485. [Google Scholar]
- Fan, X.; Liang, J.; Wu, Z.; Shan, X.; Qiao, H.; Jiang, T. Expression of HLA-DR genes in gliomas: Correlation with clinicopathological features and prognosis. Chin. Neurosurg. J. 2017, 3, 154–162. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Li, Y.T.; Yan, T.F.; Xu, Y.; Liu, B.H.; Yang, J.A.; Yang, X.; Chen, Q.X.; Zhang, H.B. Six immune associated genes construct prognostic model evaluate low-grade glioma. Front. Immunol. 2020, 11, 606164. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, L.; Wang, Z.; Yang, F.; Wang, H.; Liang, T.; Wu, F.; Lan, Q.; Wang, J.; Zhao, J. A three-gene signature for prognosis in patients with MGMT promoter-methylated glioblastoma. Oncotarget 2016, 7, 69991. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Ren, X.; Zhang, C.; Cai, J.; Liu, Y.; Han, S.; Wu, A. Bioinformatic profiling identifies an immune-related risk signature for glioblastoma. Neurology 2016, 86, 2226–2234. [Google Scholar] [CrossRef]
- Menyhárt, O.; Fekete, J.T.; Győrffy, B. Gene expression-based biomarkers designating glioblastomas resistant to multiple treatment strategies. Carcinogenesis 2021, 42, 804–813. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Peng, Q.; Li, R.; Li, Y.; Xu, X.; Ni, W.; Lin, H.; Ning, L. Prediction of a competing endogenous RNA co-expression network as a prognostic marker in glioblastoma. J. Cell. Mol. Med. 2020, 24, 13346–13355. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef]
- Cioca, A.; Olteanu, E.G.; Gisca, M.D.; Morosanu, C.O.; Marin, I.; Florian, I.S. Expression of EGFR in paired new and recurrent glioblastomas. Asian Pac. J. Cancer Prev. 2016, 17, 4205–4208. [Google Scholar]
- Jensen, K.V.; Hao, X.; Aman, A.; Luchman, H.A.; Weiss, S. EGFR blockade in GBM brain tumor stem cells synergizes with JAK2/STAT3 pathway inhibition to abrogate compensatory mechanisms in vitro and in vivo. Neuro-Oncol. Adv. 2020, 2, vdaa020. [Google Scholar] [CrossRef]
- Dai, X.; Ye, L.; Cheng, H. Interaction between microglial cells and neural stem cells influences the relapse of glioblastoma. Brain Tumor Res. Treat. 2022, 10, S265. [Google Scholar]
- Dréan, A.; Goldwirt, L.; Verreault, M.; Canney, M.; Schmitt, C.; Guehennec, J.; Delattre, J.Y.; Carpentier, A.; Idbaih, A. Blood-brain barrier, cytotoxic chemotherapies and glioblastoma. Expert Rev. Neurother. 2016, 16, 1285–1300. [Google Scholar] [CrossRef]
- Roy, L.O.; Lemelin, M.; Blanchette, M.; Poirier, M.B.; Aldakhil, S.; Fortin, D. Expression of ABCB1, ABCC1 and 3 and ABCG2 in glioblastoma and their relevance in relation to clinical survival surrogates. J. Neuro-Oncol. 2022, 160, 601–609. [Google Scholar] [CrossRef]
- Ting, J.P.Y.; Trowsdale, J. Genetic control of MHC class II expression. Cell 2002, 109, S21–S33. [Google Scholar] [CrossRef]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef]
- Qian, J.; Luo, F.; Yang, J.; Liu, J.; Liu, R.; Wang, L.; Wang, C.; Deng, Y.; Lu, Z.; Wang, Y.; et al. TLR2 Promotes Glioma Immune Evasion by Downregulating MHC Class II Molecules in Microglia. Cancer Immunol. Res. 2018, 6, 1220–1233. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Jiang, Y.; Tao, D.; Wang, Z.; Wang, R.; Wang, M.; Han, S. NFAT2-HDAC1 signaling contributes to the malignant phenotype of glioblastoma. Neuro-Oncol. 2020, 22, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, H.; He, L.; Xiang, Y.; Tian, C.; Li, C.; Tan, P.; Jing, J.; Tian, Y.; Du, L.; et al. Discovery of small-molecule inhibitors of the HSP90-calcineurin-NFAT pathway against glioblastoma. Cell Chem. Biol. 2019, 26, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Shen, G.; Su, Z.; Du, J.; Xu, F.; Yu, Y. RAD21 inhibited transcription of tumor suppressor MIR4697HG and led to glioma tumorigenesis. Biomed. Pharmacother. 2020, 123, 109759. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’andolfi, M.; del Bianco, P.; Lollo, G.; Benoit, J.P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immunother. Cancer 2019, 7, 58. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef]
- Ha, E.T.; Antonios, J.P.; Soto, H.; Prins, R.M.; Yang, I.; Kasahara, N.; Liau, L.M.; Kruse, C.A. Chronic inflammation drives glioma growth: Cellular and molecular factors responsible for an immunosuppressive microenvironment. Neuroimmunol. Neuroinflammation 2014, 1, 66–76. [Google Scholar]
- Richards, L.M.; Whitley, O.K.; MacLeod, G.; Cavalli, F.M.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M.; et al. Gradient of developmental and injury response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat. Cancer 2021, 2, 157–173. [Google Scholar] [CrossRef]
- Baratta, M.G. Glioblastoma is ‘hot’ for personalized vaccines. Nat. Rev. Cancer 2019, 19, 129. [Google Scholar] [CrossRef]
- Agliardi, G.; Liuzzi, A.R.; Hotblack, A.; de Feo, D.; Núñez, N.; Stowe, C.L.; Friebel, E.; Nannini, F.; Rindlisbacher, L.; Roberts, T.A.; et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat. Commun. 2021, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Edvardsson, U.; Bergström, M.; Alexandersson, M.; Bamberg, K.; Ljung, B.; Dahllöf, B. Rosiglitazone (BRL49653), a PPARγ-selective agonist, causes peroxisome proliferator-like liver effects in obese mice. J. Lipid Res. 1999, 40, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Simonelli, F.; Li, X.; Spinello, A.; Laporte, S.; Torre, V.; Magistrato, A. Molecular Mechanisms of the Blockage of Glioblastoma Motility. J. Chem. Inf. Model. 2021, 61, 2967–2980. [Google Scholar] [CrossRef]
- Yadavalli, S.; Yenugonda, V.M.; Kesari, S. Repurposed drugs in treating glioblastoma multiforme: Clinical trials update. Cancer J. 2019, 25, 139–146. [Google Scholar] [CrossRef]
- Hughes, R.E.; Elliott, R.J.; Dawson, J.C.; Carragher, N.O. High-content phenotypic and pathway profiling to advance drug discovery in diseases of unmet need. Cell Chem. Biol. 2021, 28, 338–355. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Z.; Xia, C.; Niu, C.; Zhou, W. Non-Coding RNAs in glioma microenvironment and angiogenesis. Front. Mol. Neurosci. 2021, 14, 763610. [Google Scholar] [CrossRef]
- Mohme, M.; Schliffke, S.; Maire, C.L.; Rünger, A.; Glau, L.; Mende, K.C.; Matschke, J.; Gehbauer, C.; Akyüz, N.; Zapf, S.; et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-infiltrating Lymphocytes Immunophenotyping of T Cells in GBM. Clin. Cancer Res. 2018, 24, 4187–4200. [Google Scholar] [CrossRef] [PubMed]
- Hutóczki, G.; Bognár, L.; Tóth, J.; Scholtz, B.; Zahuczky, G.; Hanzély, Z.; Csősz, É.; Reményi-Puskár, J.; Kalló, G.; Hortobágyi, T.; et al. Effect of concomitant radiochemotherapy on invasion potential of glioblastoma. Pathol. Oncol. Res. 2016, 22, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Hudson, A.L.; Parker, N.R.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Chen, J.; Wheeler, H.R.; Howell, V.M. Glioblastoma recurrence correlates with increased APE1 and polarization toward an immuno-suppressive microenvironment. Front. Oncol. 2018, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Li, G.; Gao, J.; Sun, Y.; Liu, P.; Gao, H.; Li, P.; Lei, T.; Chen, Y.; Cheng, Y.; et al. SPOCK1 is upregulated in recurrent glioblastoma and contributes to metastasis and Temozolomide resistance. Cell Prolif. 2016, 49, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Hasan, T.; Caragher, S.P.; Shireman, J.M.; Park, C.H.; Atashi, F.; Baisiwala, S.; Lee, G.; Guo, D.; Wang, J.Y.; Dey, M.; et al. Interleukin-8/CXCR2 signaling regulates therapy-induced plasticity and enhances tumorigenicity in glioblastoma. Cell Death Dis. 2019, 10, 292. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Identifier | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | P11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial | P1_I | P2_I | P4_I | P5_I | P6_I | P7_I | P8_I | P11_I | |||
| Recurrent | P1_R | P2_R | P3_R | P4_R | P5_R | P6_R | P7_R | P9_R | P10_R | P11_R | |
| Sex | F | F | M | M | M | M | M | F | M | M | M |
| Age at diagnosis | 56 | 55 | 35 | 54 | 33 | 64 | 45 | 55 | 36 | 61 | 45 |
| Time to relapse | 462 | 266 | 312 | 511 | 336 | 541 | 658 | 348 | 395 | 343 | 427 |
| Overall survival | 757 | 454 | 449 | 764 | 493 | 588 | 987 | 651 | 751 | 665 | 556 |
| CCRT (weeks) | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| TMZ (cycles) | 6 | 5 | 6 | 6 | 3 | 0 | 2 | 6 | 4 | 6 | 6 |
| IDH1 (R132H) IHC | NEGATIVE | NEGATIVE | NEGATIVE | NEGATIVE | NEGATIVE | NEGATIVE | NEGATIVE | NEGATIVE | NEGATIVE | NEGATIVE | NOT PERFORMED |
| ATRX IHC | NEGATIVE (TI) | RETAINED | NEGATIVE (TI) | RETAINED | NEGATIVE (TI) | RETAINED | NEGATIVE (TI) | RETAINED | NEGATIVE (TI) | RETAINED | RETAINED |
| TERT | FAILED | NOT REPORTED | C250T MUTATION | FAILED | FAILED | NOT REPORTED | FAILED | FAILED | FAILED | C228T MUTATION | NOT PERFORMED |
| Histone H3F3A | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NOT PERFORMED |
| IDH1/2 Seq | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION | NO MUTATION |
| 1p/19q co-deletion | RETAINED | RETAINED | RETAINED | RETAINED * | RETAINED | RETAINED | RETAINED * | RETAINED * | RETAINED | RETAINED | RETAINED |
| EGFR | NO AMP | NO AMP | NO AMP | AMPLIFICATION | NO AMP | AMPLIFICATION | NO AMP | AMPLIFICATION | AMPLIFICATION | NO AMP | AMP AND VIII MUTATION |
| MGMT promoter | NO/INSIGNIFICANT | NO/INSIGNIFICANT | NO/INSIGNIFICANT | NO/INSIGNIFICANT | NO/INSIGNIFICANT | LOW 5–10% | INTERMEDIATE 10–25% | NO/INSIGNIFICANT | NO/INSIGNIFICANT | INTERMEDIATE 10–25% | LOW 5–10% |
| Diagnosis | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT | GBM, IDH-WT |
| Initial GEO Accession | GSM6508723 | GSM6508725 | GSM6508728 | GSM6508730 | GSM6508732 | GSM6508734 | GSM6508736 | GSM6508739 | |||
| Recurrent GEO Accession | GSM6508724 | GSM6508726 | GSM6508727 | GSM6508729 | GSM6508731 | GSM6508733 | GSM6508735 | GSM6508737 | GSM6508738 | GSM6508740 |
| FDA-Approved Drug | Description | No. of Profiles | Z-Score | Connection Score | p-Value | Cell Line * |
|---|---|---|---|---|---|---|
| nizatidine | Histamine H2-receptor antagonists | 4 | −5.376 | −0.278 | 8.19 × 10−8 | NPC |
| pantoprazole | Proton pump inhibitor | 4 | −4.458 | −0.234 | 7.37 × 10−6 | NPC |
| tolmetin | Nonsteroidal anti-inflammatory drug | 3 | −4.440 | −0.301 | 9.66 × 10−6 | NPC |
| bicalutamide | Anti-androgen drug | 4 | −4.250 | −0.254 | 1.99 × 10−5 | NPC |
| clomifene | Effective inhibitor of mutant IDH1 | 4 | −4.076 | −0.240 | 4.87 × 10−5 | NPC |
| dinoprostone | Naturally occurring prostaglandin | 10 | −3.948 | −0.145 | 7.40 × 10−5 | NPC |
| levocabastine | H1 receptor antagonist | 2 | −3.929 | −0.293 | 7.51 × 10−5 | NEU |
| hydroxychloroquine | Regulates the immune system | 6 | −4.060 | −0.189 | 4.98 × 10−5 | NEU |
| progesterone | Endogenous steroid hormone | 5 | −4.189 | −0.228 | 2.80 × 10−5 | NEU.KCL |
| gemfibrozil | Lipid-lowering drug | 6 | −4.390 | −0.193 | 1.01 × 10−5 | NEU |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roddy, A.C.; McInerney, C.E.; Flannery, T.; Healy, E.G.; Stewart, J.P.; Spence, V.J.; Walsh, J.; Salto-Tellez, M.; McArt, D.G.; Prise, K.M. Transcriptional Profiling of a Patient-Matched Cohort of Glioblastoma (IDH-Wildtype) for Therapeutic Target and Repurposing Drug Identification. Biomedicines 2023, 11, 1219. https://doi.org/10.3390/biomedicines11041219
Roddy AC, McInerney CE, Flannery T, Healy EG, Stewart JP, Spence VJ, Walsh J, Salto-Tellez M, McArt DG, Prise KM. Transcriptional Profiling of a Patient-Matched Cohort of Glioblastoma (IDH-Wildtype) for Therapeutic Target and Repurposing Drug Identification. Biomedicines. 2023; 11(4):1219. https://doi.org/10.3390/biomedicines11041219
Chicago/Turabian StyleRoddy, Aideen C., Caitríona E. McInerney, Tom Flannery, Estelle G. Healy, James P. Stewart, Veronica J. Spence, Jamie Walsh, Manuel Salto-Tellez, Darragh G. McArt, and Kevin M. Prise. 2023. "Transcriptional Profiling of a Patient-Matched Cohort of Glioblastoma (IDH-Wildtype) for Therapeutic Target and Repurposing Drug Identification" Biomedicines 11, no. 4: 1219. https://doi.org/10.3390/biomedicines11041219
APA StyleRoddy, A. C., McInerney, C. E., Flannery, T., Healy, E. G., Stewart, J. P., Spence, V. J., Walsh, J., Salto-Tellez, M., McArt, D. G., & Prise, K. M. (2023). Transcriptional Profiling of a Patient-Matched Cohort of Glioblastoma (IDH-Wildtype) for Therapeutic Target and Repurposing Drug Identification. Biomedicines, 11(4), 1219. https://doi.org/10.3390/biomedicines11041219