
c-Myc/microRNA-17-92 Axis Phase-Dependently Regulates PTEN and p21 Expression via ceRNA during Reprogramming to Mouse Pluripotent Stem Cells
 and
and 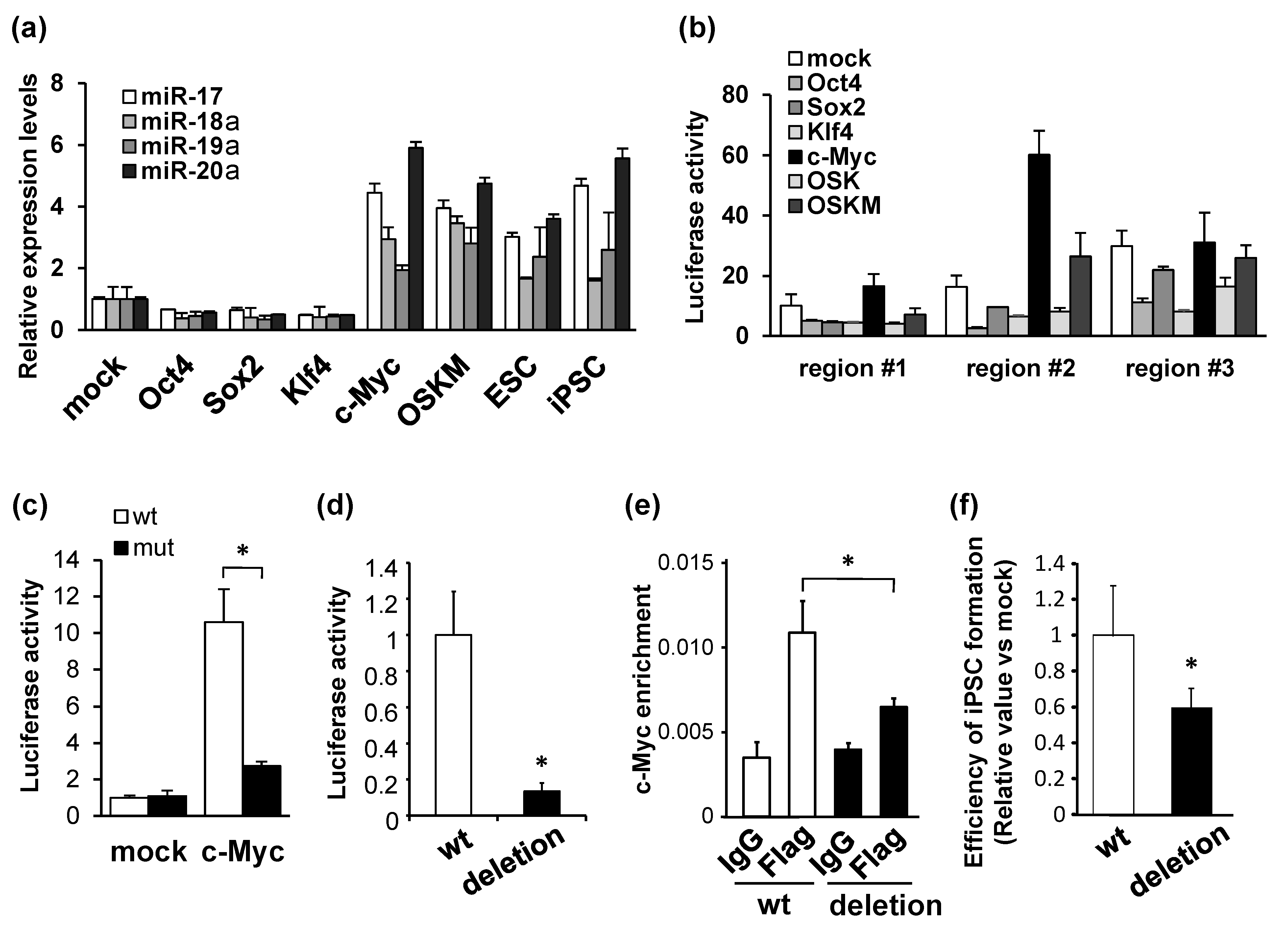{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and Lentivirus-Mediated Genome Editing
2.2. Cell Culture and Mouse iPSC Formation Experiments
2.3. Quantitative Real-Time PCR
2.4. Luciferase Reporter Assay
2.5. Chromatin Immunoprecipitation
2.6. Immunocytochemistry
2.7. In Silico Analysis
2.8. Western Blotting
2.9. Animals and Ethics
2.10. Statistics
3. Results
3.1. Activation of miR-17-92 Transcription by c-Myc
3.2. miR-17-92 Is Involved in Mouse iPSC Formation
3.3. Potential Targets of Individual miRNAs Comprising miR-17-92
3.4. PTEN and p21 Are Downregulated by miR-17 and miR-20a
3.5. iPSC Production Is Downregulated by PTEN ceRNA against miR-17-92
3.6. Post-Transcriptional Regulation of PTEN and p21 by miR-17-92 in the Context of ceRNA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [Green Version]
- Judson, R.L.; Babiarz, J.E.; Venere, M.; Blelloch, R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat. Biotechnol. 2009, 27, 459–461. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, C.S.; Nakashima, K.; Rana, T.M. Small RNA-mediated regulation of iPS cell generation. EMBO J. 2011, 30, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs-microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Park, J.E.; Heo, I.; Tian, Y.; Simanshu, D.K.; Chang, H.; Jee, D.; Patel, D.J.; Kim, V.N. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature 2011, 475, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Chopp, M.; Wang, X.L.; Zhang, L.; Hozeska-Solgot, A.; Tang, T.; Kassis, H.; Zhang, R.L.; Chen, C.; Xu, J.; et al. MicroRNA-17-92 cluster mediates the proliferation and survival of neural progenitor cells after stroke. J. Biol. Chem. 2013, 288, 12478–12488. [Google Scholar] [CrossRef] [Green Version]
- Jouneau, A.; Ciaudo, C.; Sismeiro, O.; Brochard, V.; Jouneau, L.; Vandormael-Pournin, S.; Coppee, J.Y.; Zhou, Q.; Heard, E.; Antoniewski, C.; et al. Naive and primed murine pluripotent stem cells have distinct miRNA expression profiles. RNA 2012, 18, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Izpisua Belmonte, J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.S.; Kawamura, T.; Wei, Z.; Sogo, T.; Jacinto, S.; Shigeno, A.; Kushige, H.; Yoshihara, E.; Liddle, C.; Ecker, J.R.; et al. ERRs mediate a metabolic switch required for somatic cell reprogramming to pluripotency. Cell Stem Cell 2015, 16, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Marumoto, T.; Yamaguchi, S.; Okano, S.; Takeda, N.; Sakamoto, C.; Kawano, H.; Nii, T.; Miyamato, S.; Nagai, Y.; et al. Inhibition of PTEN tumor suppressor promotes the generation of induced pluripotent stem cells. Mol. Ther. 2013, 21, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, M.; Kimura, T. RNA networks that regulate mRNA expression and their potential as drug targets. RNA Dis. 2015, 3, e864. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Canamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef] [Green Version]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Pink, R.C.; Wicks, K.; Caley, D.P.; Punch, E.K.; Jacobs, L.; Carter, D.R. Pseudogenes: Pseudo-functional or key regulators in health and disease? RNA 2011, 17, 792–798. [Google Scholar] [CrossRef] [Green Version]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; DeNicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In vivo identification of tumor-suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 2011, 147, 382–395. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishida, T.; Ueyama, T.; Ihara, D.; Harada, Y.; Nakagawa, S.; Saito, K.; Nakao, S.; Kawamura, T. c-Myc/microRNA-17-92 Axis Phase-Dependently Regulates PTEN and p21 Expression via ceRNA during Reprogramming to Mouse Pluripotent Stem Cells. Biomedicines 2023, 11, 1737. https://doi.org/10.3390/biomedicines11061737
Ishida T, Ueyama T, Ihara D, Harada Y, Nakagawa S, Saito K, Nakao S, Kawamura T. c-Myc/microRNA-17-92 Axis Phase-Dependently Regulates PTEN and p21 Expression via ceRNA during Reprogramming to Mouse Pluripotent Stem Cells. Biomedicines. 2023; 11(6):1737. https://doi.org/10.3390/biomedicines11061737
Chicago/Turabian StyleIshida, Tomoaki, Tomoe Ueyama, Dai Ihara, Yukihiro Harada, Sae Nakagawa, Kaho Saito, Shu Nakao, and Teruhisa Kawamura. 2023. "c-Myc/microRNA-17-92 Axis Phase-Dependently Regulates PTEN and p21 Expression via ceRNA during Reprogramming to Mouse Pluripotent Stem Cells" Biomedicines 11, no. 6: 1737. https://doi.org/10.3390/biomedicines11061737
APA StyleIshida, T., Ueyama, T., Ihara, D., Harada, Y., Nakagawa, S., Saito, K., Nakao, S., & Kawamura, T. (2023). c-Myc/microRNA-17-92 Axis Phase-Dependently Regulates PTEN and p21 Expression via ceRNA during Reprogramming to Mouse Pluripotent Stem Cells. Biomedicines, 11(6), 1737. https://doi.org/10.3390/biomedicines11061737