Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis



Abstract
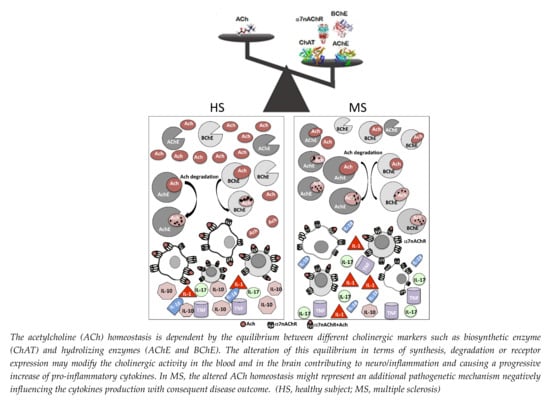
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Acetylcholine and Immune System
2. Cholinergic Dysfunction in Multiple Sclerosis
3. The Interface between Cholinergic System and Inflammatory Cytokines in MS
4. Genetic Polymorphisms for BChE and AChE and MS
5. Cholinergic Markers Alteration in the Brain of EAE Mice
6. Cholinergic Receptors in the Glial Cells
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mesulam, M.M. Central cholinergic pathways: Neuroanatomy and some behavioral implications. In Neurotransmitters and Cortical Function; Avoli, M., Reader, T.A., Dikes, R.W., Gloor, P., Eds.; Plenum Publishing: New York, NY, USA, 1988; pp. 237–260. [Google Scholar]
- Wessler, I.; Kilbinger, H.; Bittinger, F.; Unger, R.; Kirkpatrick, C.J. The biological role of non-neuronal acetylcholine in plants and humans. Jpn. J. Pharmacol. 2001, 85, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, K.; Fujii, T. Basic and clinical aspects of non-neuronal acetylcholine: Overview of non-neuronal cholinergic systems and their biological significance. J. Pharmacol. Sci. 2008, 106, 167–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furchgott, R.F.; Carvalho, M.H.; Khan, M.T.; Matsunaga, K. Evidence for endothelium-dependent vasodilation of resistance vessels by acetylcholine. Blood Vessel. 1987, 24, 145–149. [Google Scholar] [CrossRef]
- Grando, S.A.; Pittelkow, M.R.; Schellreunter, K.U. Adrenergic and cholinergic control in the biology of epidermis: Physiological and clinical Significance. J. Invest. Dermatol. 2006, 126, 1948–1965. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.A.; Bassi, C.; Saunders, M.E.; Nechanitzky, R.; Morgado-Palacin, I.; Zheng1, C.; Mak, T.W. Beyond neurotransmission: Acetylcholine in immunity and inflammation. J. Int. Med. 2020, 287, 120–133. [Google Scholar] [CrossRef]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K.; Kawashima, K. Physiological functions of the cholinergic system in immune cells. J. Pharmacol. Sci. 2017, 134, 1–21. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- Carnevale, D.; Perrotta, M.; Pallante, F.; Fardella, V.; Iacobucci, R.; Fardella, S.; Carnevale, L.; Carnevale, R.; de Lucia, M.; Cifelli, G.; et al. A cholinergic sympathetic pathway primes immunity in hypertension and mediates brain-to-spleen communication. Nat. Commun. 2016, 7, 13035. [Google Scholar] [CrossRef] [Green Version]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdés-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Soreq, H.; Seidman, S. Acetylcholinesterase–new roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Fujii, T. The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci. 2003, 74, 675–696. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K.; Kawashima, K. Expression and Function of the Cholinergic System in Immune Cells. Front. Immunol. 2017, 8, 1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, H.; Fujii, T.; Watanabe, Y.; Kawashima, K. Expression of multiple mRNA species for choline acetyltransferase in human T-lymphocytes. Life Sci. 2003, 72, 2127–2130. [Google Scholar] [CrossRef]
- Kawashima, K.; Fujii, T. Extraneuronal cholinergic system in lymphocytes. Pharmacol. Ther. 2000, 86, 29–48. [Google Scholar] [CrossRef]
- Tata, A.M. Muscarinic acetylcholine receptors: New potential therapeutic targets in antinociception and in cancer therapy. Rec Pat. CNS Drug Discov. 2008, 3, 94–103. [Google Scholar] [CrossRef]
- Kawashima, K.; Fujii, T.; Watanabe, Y.; Misawa, H. Acetylcholine synthesis and muscarinic receptor subtype mRNA expression in T-lymphocytes. Life Sci. 1998, 62, 1701–1705. [Google Scholar] [CrossRef]
- Fujii, T.; Watanabe, Y.; Inoue, T.; Kawashima, K. Upregulation of mRNA encoding the M5 muscarinic acetylcholine receptor in human T- and B-lymphocytes during immunological responses. Neurochem. Res. 2003, 28, 423–429. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor [alpha] 7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Skok, M.V.; Grathe, K.; Agenes, F.; Changeux, P.J. The role of nicotinic receptorsi in B-lymphocites development and activation. Life Sci. 2007, 80, 2324–2336. [Google Scholar] [CrossRef]
- Kawashima, K.; Fuji, T.; Moriwaki, Y.; Misawa, H.; Horiguchi, K. Non-neuronal cholinergic system in regulation of immune function with a focus on α7 nAChRs. Int. Immunopharmacol. 2015, 29, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizri, E.; Hamra-Amitay, Y.; Sicsic, C.; Lavon, I.; Brenner, T. Anti-inflammatory properties of cholinergic up-regulation: A new role for acetylcholinesterase inhibitors. Neuropharmacology 2006, 50, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Nirzi, E.; Jromy-Tir-Sinai, M.; Faranesh, N.; Layon, I.; Lavi, E.; Weinstock, M.; Brenner, T. Supprssion of neuroinflammationby the acetylcholinesterase inhibitor rivastigamine. J. Neuroimmunol. 2008, 203, 12–22. [Google Scholar]
- Nirzi, E.; Brenner, T. Modulation of inflammatory pathways by the immune-cholinergic system. Amino Acids 2013, 45, 75–85. [Google Scholar]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J. Immunol. 2010, 183, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reale, M.; De Angelis, F.; Di Nicola, M.; Capello, E.; Di Ioia, M.; De Luca, G.; Lugaresi, A.; Tata, A.M. Relation Between Pro-Inflammatory Cytokines and Acetylcholine Levels in Relapsing-Remitting Multiple Sclerosis Patients. Int. J. Mol. Sci. 2012, 13, 12656–12664. [Google Scholar] [CrossRef] [Green Version]
- Di Bari, M.; Di Pinto, G.; Reale, M.; Mengod, G.; Tata, A.M. Cholinergic system and neuroinflammation: Implication in multiple sclerosis. CNS Agents Med. Chem. 2017, 17, 109–115. [Google Scholar] [CrossRef]
- Di Bari, M.; Reale, M.; Di Nicola, M.; Orlando, V.; Galizia, S.; Porfilio, I.; Costantini, E.; D’Angelo, C.; Ruggieri, S.; Biagioni, S.; et al. Dysregulated homeostasis of acetylcholine levels in immune cells of RR-multiple sclerosis patients. Int. J. Mol. Sci. 2016, 17, E2009. [Google Scholar] [CrossRef] [Green Version]
- Tata, A.M.; Velluto, L.; D’Angelo, C.; Reale, M. Cholinergic system dysfunctions and neurodegenerative diseases: Cause or effect? CNS Neurol. Disord. Drug Targets 2014, 13, 1294–1303. [Google Scholar] [CrossRef]
- Yilmaz, V.; Oflazer, P.; Aysal, F.; Durmus, H.; Poulas, K.; Yentur, S.P.; Gulsen-Parman, Y.; Tzartos, S.; Marx, A.; Tuzun, E.; et al. Differential Cytokine Changes in Patients with Myasthenia Gravis with Antibodies against AChR and MuSK. PLoS ONE 2015, 10, e0123546. [Google Scholar] [CrossRef] [Green Version]
- Baggi, F.; Antozzi, C.; Toscani, C.; Cordiglieri, C. Acetylcholine receptor-induced experimental myasthenia gravis: What have we learned from animal models after three decades? Arch. Immunol. Ther. Exp. 2012, 60, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Peña, G.; Cai, B.; Ramos, L.; Vida, G.; Deitch, E.A.; Ulloa, L. Cholinergic regulatory lymphocytes re-establish neuromodulation of innate immune responses in sepsis. J. Immunol. 2011, 187, 718–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Song, F. The Properties of Cytokines in Multiple Sclerosis: Pros and Cons. Am. J. Med. Sci. 2018, 356, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Di Bari, M.; Di Nicola, M.; D’Angelo, C.; De Angelis, F.; Velluto, L.; Tata, A.M. Nicotinic receptor activation negatively modulates pro-inflammatory cytokine production in multiple sclerosis patients. Int. Immunopharmacol. 2015, 29, 152–157. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, C.; Reale, M. IFN-β-Treatment and Canonical and Non-Traditional Cytokine Levels. Front. Immunol. 2018, 9, 1240. [Google Scholar] [CrossRef] [PubMed]
- Báez-Pagán, C.A.; Delgado-Vélez, M. Activation of the Macrophage α7 Nicotinic Acetylcholine Receptor and Control of Inflammation. J. Neuroimmune Pharmacol. 2015, 10, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Marrosu, M.G.; Lai, M.; Cocco, E.; Loi, V.; Spinicci, G.; Pischedda, M.P.; Massole, S.; Marrosu, G.; Contu, P. Genetic factors and the founder effect explain familial MS in Sardinia. Neurology 2002, 58, 283–288. [Google Scholar] [CrossRef]
- Kahana, E. Epidemiologic studies of multiple sclerosis: A review. Biomed. Pharmacother. 2000, 54, 100–112. [Google Scholar] [CrossRef]
- Carton, H.; Vlietinck, R.; Debruyne, J.; De, K.J.; D’Hooghe, M.B.; Loos, R.; Medaer, R.; Truyen, L.; Yee, I.M.; Sadovnick, A.D. Risks of multiple sclerosis in relatives of patients in Flanders, Belgium. J. Neurol. Neuros. Psych. 1997, 62, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.R. Esclerose múltipla. In Tratado de Neurologia, 10th ed.; Sociedad de Neurología: Santiago, Chile, 2002; pp. 670–686. [Google Scholar]
- Buscarinu, M.C.; Fornasiero, A.; Ferraldeschi, M.; Romano, S.; Reniè, R.; Morena, E.; Romano, C.; Pellicciari, G.; Landi, A.C.; Fagnani, C.; et al. Disentangling the molecular mechanisms of multiple sclerosis: The contribution of twin studies. Neurosci. Biobehav. Rev. 2020, 111, 194–198. [Google Scholar] [CrossRef]
- Hollenbach, J.A.; Oksenberg, J.R. The immunogenetics of multiple sclerosis: A comprehensive review. J. Autoimmun. 2015, 64, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallaur, A.P.; Kaimen-Maciel, D.R.; Morimoto, H.K.; Watanabe, M.A.; Georgeto, S.M.; Reiche, E.M. Genetic polymorphisms associated with the development and clinical course of multiple sclerosis. Int. J. Mol. Med. 2011, 28, 467–479. [Google Scholar] [PubMed]
- Ben-David, Y.; Kagan, S.; Cohen Ben-Ami, H.; Rostami, J.; Mizrahi, T.; Kulkarni, A.R.; Thakur, G.A.; Vaknin-Dembinsky, A.; Healy, L.M.; Brenner, T.; et al. RIC3, the cholinergic anti-inflammatory pathway, and neuroinflammation. Int. Immunopharmacol. 2020, 83, 106381. [Google Scholar] [CrossRef] [PubMed]
- Jasiecki, J.; Wasąg, B. Butyrylcholinesterase Protein Ends in the Pathogenesis of Alzheimer’s Disease-Could BCHE Genotyping Be Helpful in Alzheimer’s Therapy. Biomolecules 2019, 9, 592. [Google Scholar] [CrossRef] [Green Version]
- Jasiecki, J.; Limon-Sztencel, A.; Żuk, M.; Chmara, M.; Cysewski, D.; Limon, J.; Wasąg, B. Synergy between the alteration in the N-terminal region of butyrylcholinesterase K variant and apolipoprotein E4 in late-onset Alzheimer’s disease. Sci. Rep. 2019, 9, 5223. [Google Scholar] [CrossRef]
- Scacchi, R.; Ruggeri, M.; Corbo, R.M. Variation of the butyrylcholinesterase (BChE) and acetylcholinesterase (AChE) genes in coronary artery disease. Clin. Chim. Acta 2011, 412, 1341–1344. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chou, W.H.; Fang, C.P.; Liu, T.H.; Tsou, H.H.; Wang, Y.; Liu, Y.L. Serum Level and Activity of Butylcholinesterase: A Biomarker for Post-Stroke Dementia. J. Clin. Med. 2019, 8, 1778. [Google Scholar] [CrossRef] [Green Version]
- Lockridge, O.; Norgren, R.B., Jr.; Johnson, R.C.; Blake, T.A. Naturally Occurring Genetic Variants of Human Acetylcholinesterase and Butyrylcholinesterase and Their Potential Impact on the Risk of Toxicity from Cholinesterase Inhibitors. Chem. Res. Toxicol. 2016, 29, 1381–1392. [Google Scholar] [CrossRef]
- Valle, A.M.; Radic, Z.; Rana, B.K.; Mahboubi, V.; Wessel, J.; Shih, P.A.; Rao, F.; O’Connor, D.T.; Taylor, P. Naturally occurring variations in the human cholinesterase genes: Heritability and association with cardiovascular and metabolic traits. J. Pharmacol. Exp. Ther. 2011, 338, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Santarpia, L.; Grandone, I.; Contaldo, F.; Pasanisi, F. Butyrylcholinesterase as a prognostic marker: A review of the literature. J. Cachexia Sarcopenia Muscle 2013, 4, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.X.; Xu, X.M.; Hu, L.; Liu, Y.; Huang, Y.J.; Wei, Y.D. Serum Butyrylcholinesterase Activity: A Biomarker for Parkinson’s Disease and Related Dementia. BioMed. Res. Int. 2017, 2017, 1524107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguri, M.; Kato, K.; Yokoi, K.; Yoshida, T.; Watanabe, S.; Metoki, N.; Yoshida, H.; Satoh, K.; Aoyagi, Y.; Tanaka, M.; et al. Association of a polymorphism of BCHE with ischemic stroke in Japanese individuals with chronic kidney disease. Mol. Med. Rep. 2009, 2, 779–785. [Google Scholar] [PubMed] [Green Version]
- Reale, M.; Costantini, E.; Di Nicola, M.; D’Angelo, C.; Franchi, S.; D’Aurora, M.; Di Bari, M.; Orlando, V.; Galizia, S.; Ruggieri, S.; et al. Butyrylcholinesterase and Acetylcholinesterase polymorphisms in Multiple Sclerosis patients: Implication in peripheral inflammation. Sci. Rep. 2018, 8, 1319. [Google Scholar] [CrossRef] [PubMed]
- Dyment, D.A.; Cader, M.Z.; Chao, M.J.; Lincoln, M.R.; Morrison, K.M.; Disanto, G.; Morahan, J.M.; De Luca, G.C.; Sadovnick, A.D.; Lepage, P.; et al. Exome sequencing identifies a novel multiple sclerosis susceptibility variant in the TYK2 gene. Neurology 2012, 79, 406–411. [Google Scholar] [CrossRef] [Green Version]
- Ramagopalan, S.V.; Dyment, D.A.; Cader, M.Z.; Morrison, K.M.; Disanto, G.; Morahan, J.M.; Berlanga-Taylor, A.J.; Handel, A.; de Luca, G.C.; Sadovnick, D.P.A.D.; et al. Rare variants in the CYP27B1 gene are associated with multiple sclerosis. Ann. Neurol. 2011, 70, 881–886. [Google Scholar] [CrossRef]
- Jamebozorgi, K.; Rostami, D.; Pormasoumi, H.; Taghizadeh, E.; Barreto, G.E.; Sahebkar, A. Epigenetic aspects of multiple sclerosis and future therapeutic options. Int. J. Neurosci. 2020, 1–15. [Google Scholar] [CrossRef]
- Waubant, E.; Lucas, R.; Mowry, E.; Graves, J.; Olsson, T.; Alfredsson, L.; Langer-Gould, A. Environmental and genetic risk factors for MS: An integrated review. Ann. Clin. Transl. Neurol. 2019, 9, 1905–1922. [Google Scholar] [CrossRef]
- Swanborg, R.H. Experimental autoimmune encephalomyelitis in rodents as a model for human demyelinating disease. Clin. Immunol. Immunopathol. 1995, 77, 4–13. [Google Scholar] [CrossRef]
- Brown, D.A.; Sawchenko, P.E. Time course and distribution of inflammatory and neurodegenerative events suggest structural bases for the pathogenesis of experimental autoimmune encephalomyelitis. J. Comp. Neurol. 2007, 502, 236–260. [Google Scholar] [CrossRef]
- Soulika, A.M.; Lee, E.; McCauley, E.; Miers, L.; Bannerman, P.; Pleasure, D. Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J. Neurosci. 2009, 29, 14965–14979. [Google Scholar] [CrossRef] [Green Version]
- Di Pinto, G.; Di Bari, M.; Martin-Alvarez, R.; Sperduti, S.; Serrano-Acedo, S.; Gatta, V.; Tata, A.M.; Mengod, G. Comparative study of the expression of cholinergic system components in the CNS of experimental autoimmune encephalomyelitis mice: Acute vs. remitting phase. Eur. J. Neurosci. 2018, 48, 2165–2181. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.I.; Geula, C.; Mesulam, M.M. Neuroglial cholinesterases in the normal brain and in Alzheimer’s disease: Relationship to plaques, tangles, and patterns of selective vulnerability. Ann. Neurol. 1993, 34, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; St Pierre, S.; Roy, P.; Morley, B.J.; Hao, J.; Simard, A.R. Infiltration of CCR2+Ly6Chigh Proinflammatory Monocytes and Neutrophils into the Central Nervous System Is Modulated by Nicotinic Acetylcholine Receptors in a Model of Multiple Sclerosis. J. Immunol. 2016, 196, 2095–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taveggia, C.; Feltri, M.L.; Wrabetz, L. Signals to promote myelin formation and repair. Nat. Rev. Neurol. 2010, 6, 276–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karadottir, R.; Attwell, D. Neurotrasmitter receptors in the life and death of oligodendrocytes. Neuroscience 2007, 145, 1426–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsetti, V.; Mozzetta, C.; Biagioni, S.; Augusti Tocco, G.; Tata, A.M. Acetylcholine release: The mechanisms and the site of release during chick dorsal root ganglia ontogenesis. Life Sci. 2012, 91, 783–788. [Google Scholar] [CrossRef]
- Bernardini, N.; Srubek Tomassy, G.; Tata, A.M.; Augusti Tocco, G.; Biagioni, S. Detection of basal and potassium-evoked acetylcholine release from embryonic DRG explants. J. Neurochem. 2004, 88, 1533–1539. [Google Scholar] [CrossRef]
- Jamebozorgi, K.; Rostami, D.; Pormasoumi, H.; Taghizadeh, E.; Barreto, G.E.; Sahebkar, A. Cholinergic signaling in myelination. Glia 2017, 65, 687–698. [Google Scholar]
- Magnaghi, V.; Procacci, P.; Tata, A.M. Novel pharmacological approaches to Schwann cells as neuroprotective agents for peripheral nerve regeneration. Int. Rev. Neurobiol. 2009, 87, 295–315. [Google Scholar]
- Larocca, J.N.; Almazan, G. Acetylcholine Agonists Stimulate Mitogen-Activated Protein Kinase in Oligodendrocyte Progenitors by Muscarinic Receptors. J. Neurosci. Res. 1997, 50, 743–754. [Google Scholar] [CrossRef]
- Ragheb, F.; Molina-Holgado, E.; Cui, Q.L.; Khorchid, A.; Liu, H.N.; Larocca, J.N.; Almazan, G. Pharmacological and functional characterization of muscarinic receptor subtypes in developing oligodendrocytes. J. Neurochem. 2001, 77, 1396–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Holgado, E.; Khorchid, A.; Liu, H.; Almazan, G. Regulation of muscarinic receptor function in developing oligodendrocytes by agonist exposure. Br. J. Pharmacol. 2003, 138, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Angelis, F.; Bernardo, A.; Magnaghi, V.; Minghetti, L.; Tata, A.M. Muscarinic receptor subtypes as potential targets to modulate oligodendrocyte progenitor survival, proliferation and differentiation. Dev. Neurobiol. 2012, 72, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. The regenerative approaches for the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, K.K.; Nissen, J.C.; Pretory, A.; Tsirka, S.E. Tuftis combines with remyelinating therapy and improves outcomes in models of CNS demyelinating disease. Front. Immunol. 2018, 9, 2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, L.; Schousboe, I.; Hertz, L.; Schousboe, A. Receptor expression in primary cultures of neurons or astrocytes. Prog Neuropsychopharmacol. Biol. Psych. 1984, 8, 521–527. [Google Scholar] [CrossRef]
- Lykhmus, O.; Voytenko, L.P.; Lips, K.S.; Bergen, I.; Krasteva-Christ, G.; Vetter, D.E.; Kummer, W.; Skok, M. Nicotinic acetylcholine receptor α9 and α10 subunits are express in the brain of mice. Front. Cell Neurosci. 2017, 11, 282. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lao, K.; Qiu, Z.; Rahman, M.S.; Zhang, Y.; Gou, X. Potential Astrocytic receptors and transporters in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 1109–1122. [Google Scholar] [CrossRef]
- Kume, T.; Takada-Takatori, Y. Nicotinic acetylcholine receptor signaling: Roles in neuroprotection. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Akaike, A., Shimohama, S., Misu, Y., Eds.; Springer: Singapore, 2018. [Google Scholar]
- Zhang, L.; McLarnon, J.G.; Goghari, V.; Lee, Y.B.; Kim, S.U.; Krieger, C. Cholinergic agonists increase intracellular Ca2+ in cultured human microglia. Neurosci. Lett. 1998, 255, 33–36. [Google Scholar] [CrossRef]
- Pannell, M.; Meier, M.A.; Szulzewsky, F.; Matyash, V.; Endres, M.; Kronenberg, G.; Prinz, V.; Waiczies, S.; Wolf, S.A.; Kettenmann, H. The subpopulation of microglia expressing functional muscarinic acetylcholine receptors expands in stroke and Alzheimer’s disease. Brain Struct. Funct. 2016, 221, 1157–1172. [Google Scholar] [CrossRef]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by a7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Chen, X.W.; Wei, W.; Liu, C. Activation of α7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through reulation of β-arrestin. Neurosci. Ther. 2017, 23, 875–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, R.; Ajimone-Cat, M.A.; Carnevale, D.; Minghetti, L. Activation of α7 nicotinic acetylcholine receptor by nicotine selectively upregulates ciclooxigenase 2 and prostaglandin E2 in rat microglial cultures. J. Neuroinflamm. 2005, 2, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; Leon, R.; Lopea, M.G. Anty inflammatory role of microglia α7 nAChRs and its role in neuroprotection. Biochem. Pharmacol. 2015, 97, 463–472. [Google Scholar] [CrossRef]
- Nicoletti, C.G.; Landi, D.; Monteleone, F.; Mataluni, G.; Albanese, M.; Lauretti, B.; Rocchi, C.; Simonelli, I.; Boffa, L.; Buttari, F.; et al. Treatment with Dimethyl Fumarate Enhances Cholinergic Transmission in Multiple Sclerosis. CNS Drugs 2019, 33, 1133–1139. [Google Scholar] [CrossRef]


 , ACh;
, ACh;  , AChE;
, AChE;  , BChE,
, BChE,  ChAT;
ChAT;  , α7nAChR.
, ACh; , AChE; , BChE, ChAT; , α7nAChR.
, α7nAChR.
, ACh; , AChE; , BChE, ChAT; , α7nAChR.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatta, V.; Mengod, G.; Reale, M.; Tata, A.M. Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis. Biomedicines 2020, 8, 153. https://doi.org/10.3390/biomedicines8060153
Gatta V, Mengod G, Reale M, Tata AM. Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis. Biomedicines. 2020; 8(6):153. https://doi.org/10.3390/biomedicines8060153
Chicago/Turabian StyleGatta, Valentina, Guadalupe Mengod, Marcella Reale, and Ada Maria Tata. 2020. "Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis" Biomedicines 8, no. 6: 153. https://doi.org/10.3390/biomedicines8060153
APA StyleGatta, V., Mengod, G., Reale, M., & Tata, A. M. (2020). Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis. Biomedicines, 8(6), 153. https://doi.org/10.3390/biomedicines8060153