Evidence of Altered Peripheral Nerve Function in a Rodent Model of Diet-Induced Prediabetes

 ,
,  , , and
, , and
Abstract
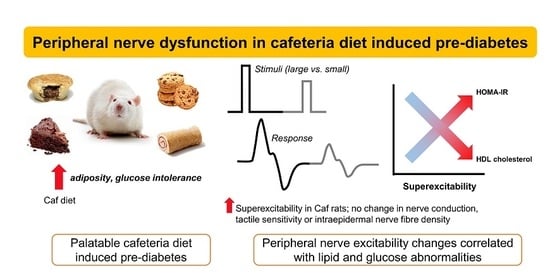
:
1. Introduction
2. Experimental Section
2.1. Animals and Diets
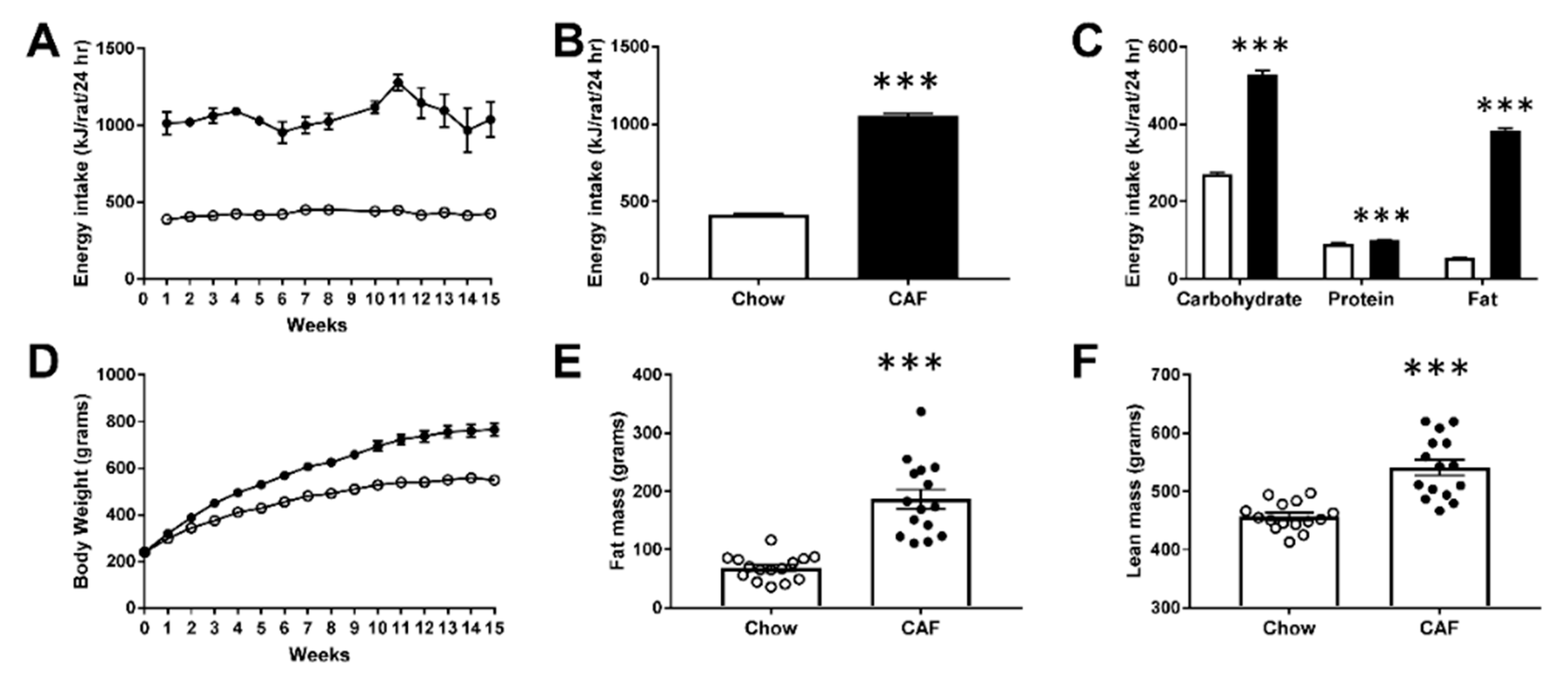
2.2. Energy Intake, Body Weight and Body Composition Analysis
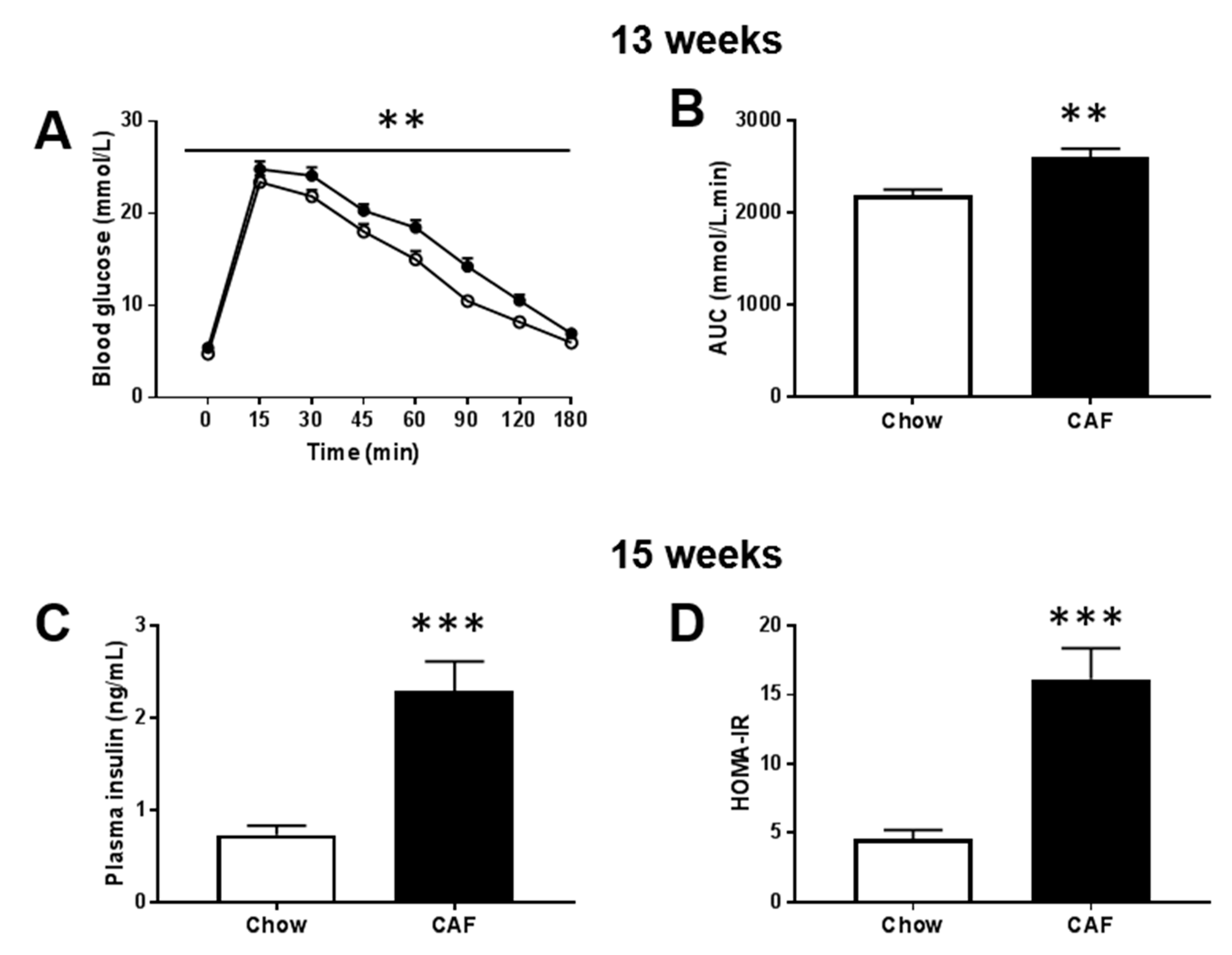
2.3. Fasting Glucose and Intraperitoneal Glucose Tolerance Test (ipGTT)
2.4. Lipids, Insulin, HOMA-Insulin Resistance, Leptin and Cytokines
2.5. Tactile Sensitivity
2.6. Nerve Conduction and Nerve Excitability Studies
2.6.1. Nerve Conduction Studies (NCS)
2.6.2. Nerve Excitability
2.6.3. Mathematical Modeling of Nerve Excitability Data
2.7. Cull and Tissue Collection
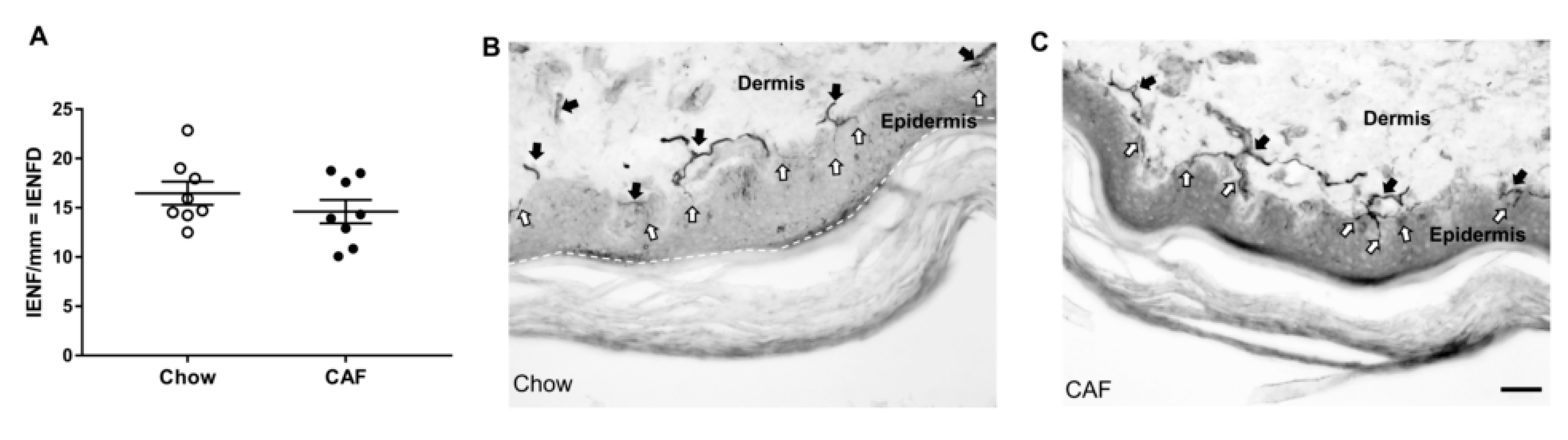
2.8. Foot Pad Intraepidermal Nerve Fiber Density (IENFD) Analysis
2.9. Statistical Analyses
3. Results
3.1. Metabolic Phenotype
3.2. Peripheral Nerve Function and Neuropathy Phenotype
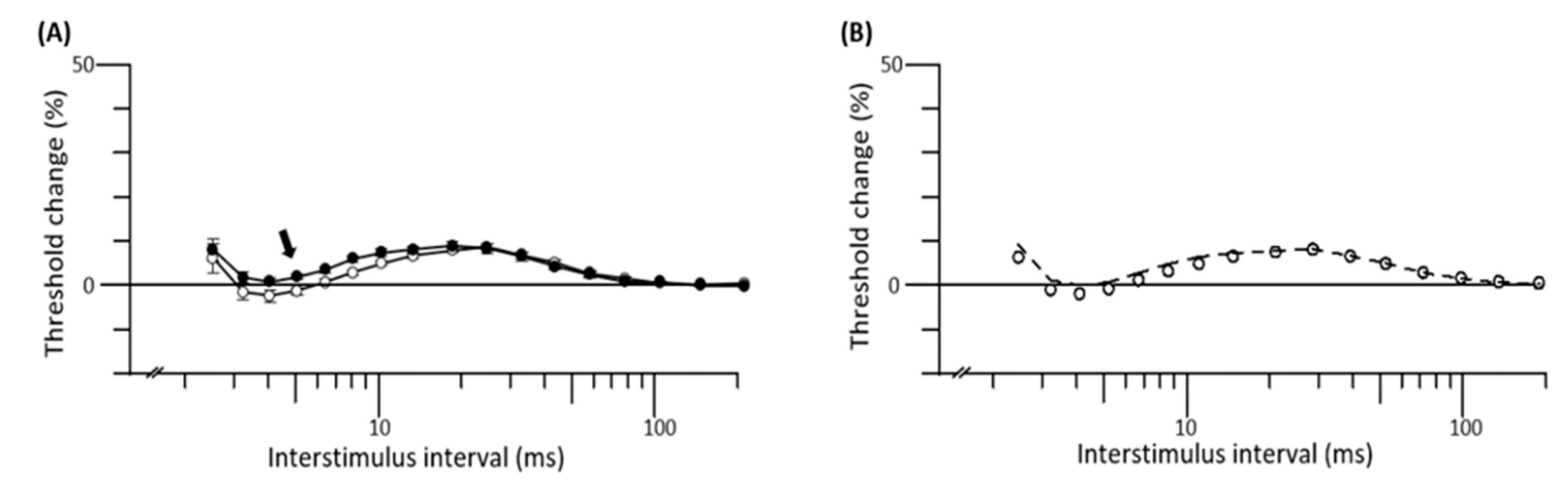
3.3. Mathematical Modelling of Nerve Excitability
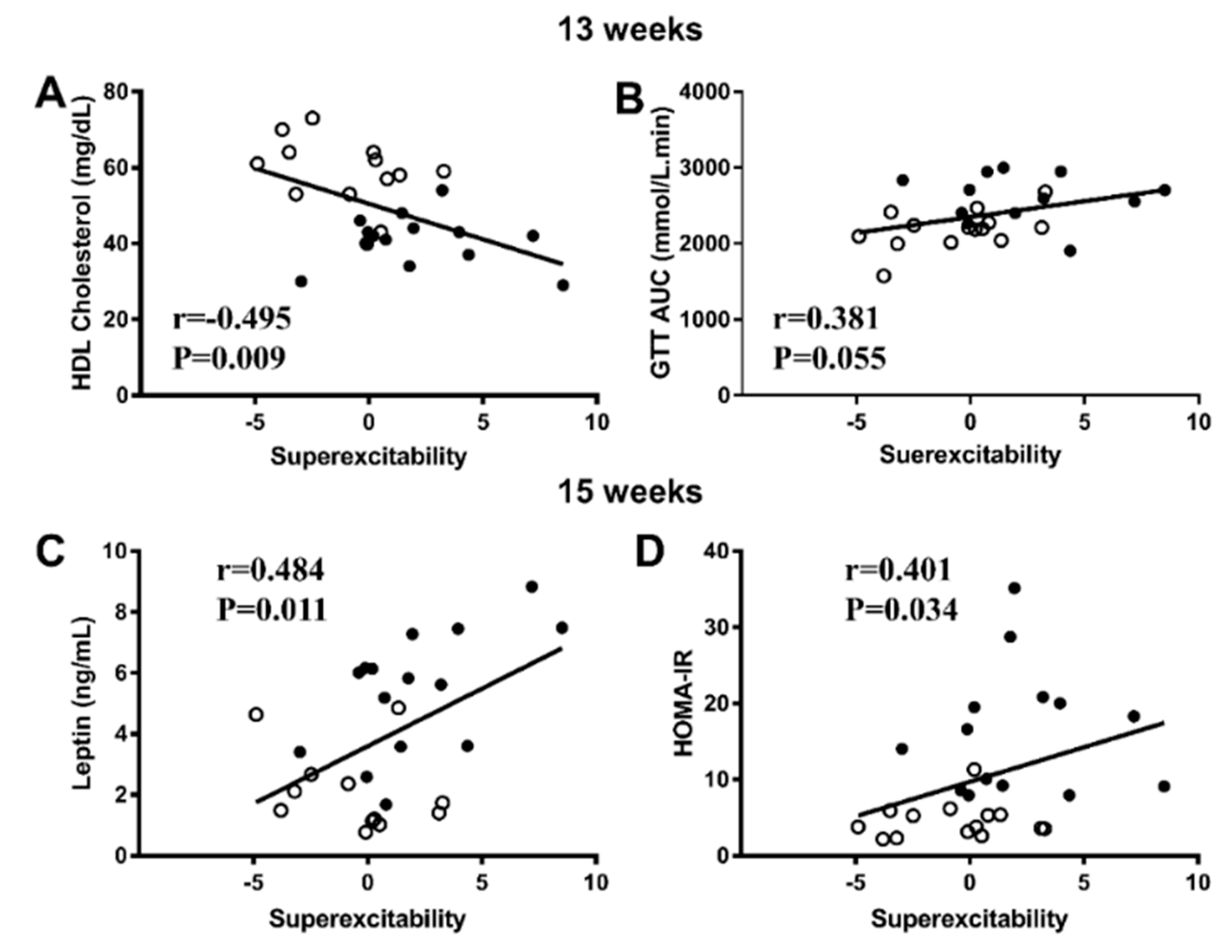
3.4. Correlations between Nerve Parameters and Metabolic Measures
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF diabetes atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Boulton, A.J.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic neuropathy: A position statement by the american diabetes association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Callaghan, B.C.; Gallagher, G.; Fridman, V.; Feldman, E.L. Diabetic neuropathy: What does the future hold? Diabetologia 2020, 63, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.H.; Knudsen, S.T.; Gylfadottir, S.S.; Christensen, L.B.; Nielsen, J.S.; Beck-Nielsen, H.; Sorensen, H.T.; Andersen, H.; Callaghan, B.C.; Feldman, E.L.; et al. Metabolic factors, lifestyle habits, and possible polyneuropathy in early type 2 diabetes: A nationwide study of 5,249 patients in the danish centre for strategic research in type 2 diabetes (DD2) cohort. Diabetes Care 2020, 43, 1266–1275. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hinder, L.M.; Rumora, A.E.; Hayes, J.M.; Dauch, J.R.; Backus, C.; Mendelson, F.E.; Feldman, E.L. Juvenile murine models of prediabetes and type 2 diabetes develop neuropathy. Dis. Model Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Perkins, B.A.; Kayaniyil, S.; Harris, S.B.; Retnakaran, R.; Gerstein, H.C.; Zinman, B.; Hanley, A.J. Peripheral neuropathy and nerve dysfunction in individuals at high risk for type 2 diabetes: The promise cohort. Diabetes Care 2015, 38, 793–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stino, A.M.; Rumora, A.E.; Kim, B.; Feldman, E.L. Evolving concepts on the role of dyslipidemia, bioenergetics, and inflammation in the pathogenesis and treatment of diabetic peripheral neuropathy. J. Peripher. Nerv. Syst. 2020, 25, 76–84. [Google Scholar] [CrossRef]
- Azmi, S.; Petropoulos, I.N.; Ferdousi, M.; Ponirakis, G.; Alam, U.; Malik, R.A. An update on the diagnosis and treatment of diabetic somatic and autonomic neuropathy. F1000Res 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biessels, G.J.; Bril, V.; Calcutt, N.A.; Cameron, N.E.; Cotter, M.A.; Dobrowsky, R.; Feldman, E.L.; Fernyhough, P.; Jakobsen, J.; Malik, R.A.; et al. Phenotyping animal models of diabetic neuropathy: A consensus statement of the diabetic neuropathy study group of the EASD (Neurodiab). J. Peripher. Nerv. Syst. 2014, 19, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Rippe, J.M.; Angelopoulos, T.J. Relationship between added sugars consumption and chronic disease risk factors: Current understanding. Nutrients 2016, 8, 697. [Google Scholar] [CrossRef] [Green Version]
- Kopp, W. How western diet and lifestyle drive the pandemic of obesity and civilization diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2221–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, S.J.; Kendig, M.D.; Morris, M.J. Palatable western-style cafeteria diet as a reliable method for modeling diet.-induced obesity in rodents. J. Vis. Exp. 2019, 153. [Google Scholar] [CrossRef] [PubMed]
- Leigh, S.-J.; Kaakoush, N.O.; Bertoldo, M.J.; Westbrook, R.F.; Morris, M.J. Intermittent cafeteria diet identifies fecal microbiome changes as a predictor of spatial recognition memory impairment in female rats. Transl. Psychiatry 2020, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.J.; Jovanovska, V.; Morris, M.J. Adaptive responses in hypothalamic neuropeptide Y in the face of prolonged high-fat feeding in the rat. J. Neurochem. 2004, 88, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, M.; Alvarez, S.; Pinchenko, V.; Marklund, S.; Graffmo, K.S.; Krarup, C. Nerve excitability changes related to axonal degeneration in amyotrophic lateral sclerosis: Insights from the transgenic SOD1(G127X) mouse model. Exp. Neurol. 2012, 233, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.; Kwai, N.C.; Krishnan, A.V. Mechanisms of axonal dysfunction in diabetic and uraemic neuropathies. Clin. Neurophysiol. 2013, 124, 2079–2090. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Park, S.B.; Islam, B.; Tamburin, S.; Velasco, R.; Alberti, P.; Bruna, J.; Psimaras, D.; Cavaletti, G.; Cornblath, D.R.; et al. Neurophysiological, nerve imaging and other techniques to assess chemotherapy-induced peripheral neurotoxicity in the clinical and research settings. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.Y.; Park, S.B.; Liu, Y.T.; Kwai, N.; Arnold, R.; Krishnan, A.V.; Lin, C.S. Progressive axonal dysfunction precedes development of neuropathy in type 2 diabetes. Diabetes 2012, 61, 1592–1598. [Google Scholar] [CrossRef] [Green Version]
- Arnold, R.; Kwai, N.; Lin, C.S.; Poynten, A.M.; Kiernan, M.C.; Krishnan, A.V. Axonal dysfunction prior to neuropathy onset in type 1 diabetes. Diabetes Metab. Res. Rev. 2013, 29, 53–59. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Dixon, W.J. Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 1980, 20, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.; Moldovan, M.; Rosberg, M.R.; Krishnan, A.V.; Morris, R.; Krarup, C. Nerve excitability in the rat forelimb: A technique to improve translational utility. J. Neurosci. Methods 2017, 275, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Burke, D.; Andersen, K.V.; Bostock, H. Multiple measures of axonal excitability: A new approach in clinical testing. Muscle Nerve 2000, 23, 399–409. [Google Scholar] [CrossRef]
- Bostock, H.; Cikurel, K.; Burke, D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve 1998, 21, 137–158. [Google Scholar] [CrossRef]
- Bostock, H. FC28. 1 MEMFIT: A computer program to aid interpretation of multiple excitability measurements on human motor axons. Clin. Neurophysiol. 2006, 117, 1. [Google Scholar] [CrossRef]
- Howells, J.; Trevillion, L.; Bostock, H.; Burke, D. The voltage dependence of I(h) in human myelinated axons. J. Physiol. 2012, 590, 1625–1640. [Google Scholar] [CrossRef]
- Makker, P.G.S.; Matamala, J.M.; Park, S.B.; Lees, J.G.; Kiernan, M.C.; Burke, D.; Moalem-Taylor, G.; Howells, J. A unified model of the excitability of mouse sensory and motor axons. J. Peripher. Nerv. Syst. 2018, 23, 159–173. [Google Scholar] [CrossRef]
- Beilharz, J.E.; Kaakoush, N.O.; Maniam, J.; Morris, M.J. Cafeteria diet and probiotic therapy: Cross talk among memory, neuroplasticity, serotonin receptors and gut microbiota in the rat. Mol. Psychiatry 2018, 23, 351–361. [Google Scholar] [CrossRef]
- Lauria, G.; Cornblath, D.R.; Johansson, O.; McArthur, J.C.; Mellgren, S.I.; Nolano, M.; Rosenberg, N.; Sommer, C. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur. J. Neurol. 2005, 12, 747–758. [Google Scholar] [CrossRef]
- Yorek, M.A. Alternatives to the streptozotocin-diabetic rodent. Int. Rev. Neurobiol. 2016, 127, 89–112. [Google Scholar]
- O’Brien, P.D.; Sakowski, S.A.; Feldman, E.L. Mouse models of diabetic neuropathy. ILAR J. 2014, 54, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Ilnytska, O.; Lyzogubov, V.V.; Pavlov, I.A.; Mashtalir, N.; Nadler, J.L.; Drel, V.R. High.-fat diet induced neuropathy of pre-diabetes and obesity: Effects of "healthy" diet and aldose reductase inhibition. Diabetes 2007, 56, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, B.C.; Gao, L.; Li, Y.; Zhou, X.; Reynolds, E.; Banerjee, M.; Pop-Busui, R.; Feldman, E.L.; Ji, L. Diabetes and obesity are the main metabolic drivers of peripheral neuropathy. Ann. Clin. Transl. Neurol. 2018, 5, 397–405. [Google Scholar] [CrossRef]
- Gerkau, N.J.; Rakers, C.; Petzold, G.C.; Rose, C.R. Differential effects of energy deprivation on intracellular sodium homeostasis in neurons and astrocytes. J. Neurosci. Res. 2017, 95, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, B.; Feldman, E. The metabolic syndrome and neuropathy: Therapeutic challenges and opportunities. Ann. Neurol. 2013, 74, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, A.G.; Bostock, H.; Finnerup, N.B.; Andersen, H.; Jensen, T.S.; Gylfadottir, S.; Itani, M.; Kroigard, T.; Sindrup, S.; Tankisi, H. Detection of early motor involvement in diabetic polyneuropathy using a novel MUNE method—MScanFit MUNE. Clin. Neurophysiol. 2019, 130, 1981–1987. [Google Scholar] [CrossRef]
- Sung, J.Y.; Tani, J.; Chang, T.S.; Lin, C.S. Uncovering sensory axonal dysfunction in asymptomatic type 2 diabetic neuropathy. PLoS ONE 2017, 12, e0171223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, J.S.; Kim, S.G.; Lim, J.C.; Chung, E.J.; Kim, O.K. Peripheral nerve axon involvement in myotonic dystrophy type 1, measured using the automated nerve excitability test. J. Clin. Neurol. 2011, 7, 90–95. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, C.S.; Chang, T.S.; Lee, J.E.; Tani, J.; Chen, H.J.; Sung, J.Y. Early sensory neurophysiological changes in prediabetes. J. Diabetes Investig. 2019. [Google Scholar] [CrossRef] [Green Version]
- George, A.; Bostock, H. Multiple measures of axonal excitability in peripheral sensory nerves: An in vivo rat model. Muscle Nerve 2007, 36, 628–636. [Google Scholar] [CrossRef]
- Callaghan, B.C.; Xia, R.; Banerjee, M.; de Rekeneire, N.; Harris, T.B.; Newman, A.B.; Satterfield, S.; Schwartz, A.V.; Vinik, A.I.; Feldman, E.L.; et al. Metabolic syndrome components are associated with symptomatic polyneuropathy independent of glycemic status. Diabetes Care 2016, 39, 801–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, B.C.; Xia, R.; Reynolds, E.; Banerjee, M.; Rothberg, A.E.; Burant, C.F.; Villegas-Umana, E.; Pop-Busui, R.; Feldman, E.L. Association between metabolic syndrome components and polyneuropathy in an obese population. JAMA Neurol. 2016, 73, 1468–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanewinckel, R.; Drenthen, J.; Ligthart, S.; Dehghan, A.; Franco, O.H.; Hofman, A.; Ikram, M.A.; van Doorn, P.A. Metabolic syndrome is related to polyneuropathy and impaired peripheral nerve function: A prospective population-based cohort study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, G.L.; Mehrabyan, A.; Simmons, K.; Amandarice; Dublin, C.; Barlow, P.; Vinik, A.I. Small fiber neuropathy is associated with the metabolic syndrome. Metab. Syndr. Relat. Disord. 2005, 3, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.M.; Hayes, J.M.; McLean, L.L.; Vivekanandan-Giri, A.; Pennathur, S.; Feldman, E.L. Dyslipidemia-induced neuropathy in mice: The role of oxLDL/LOX-1. Diabetes 2009, 58, 2376–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, P.D.; Guo, K.; Eid, S.A.; Rumora, A.E.; Hinder, L.M.; Hayes, J.M.; Mendelson, F.E.; Hur, J.; Feldman, E.L. Integrated lipidomic and transcriptomic analyses identify altered nerve triglycerides in mouse models of prediabetes and type 2 diabetes. Dis. Model Mech. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Davidson, E.P.; Coppey, L.J.; Kardon, R.H.; Yorek, M.A. Differences and similarities in development of corneal nerve damage and peripheral neuropathy and in diet-induced obesity and type 2 diabetic rats. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1222–1230. [Google Scholar] [CrossRef]
- Coppey, L.; Davidson, E.; Shevalye, H.; Torres, M.E.; Yorek, M.A. Effect of dietary oils on peripheral neuropathy-related endpoints in dietary obese rats. Diabetes Metab. Syndr. Obes. 2018, 11, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Lupachyk, S.; Watcho, P.; Obrosov, A.A.; Stavniichuk, R.; Obrosova, I.G. Endoplasmic reticulum stress contributes to prediabetic peripheral neuropathy. Exp. Neurol. 2013, 247, 342–348. [Google Scholar] [CrossRef]
- Rumora, A.E.; LoGrasso, G.; Hayes, J.M.; Mendelson, F.E.; Tabbey, M.A.; Haidar, J.A.; Lentz, S.I.; Feldman, E.L. The divergent roles of dietary saturated and monounsaturated fatty acids on nerve function in murine models of obesity. J. Neurosci. 2019, 39, 3770–3781. [Google Scholar] [CrossRef] [Green Version]
- Rumora, A.E.; Lentz, S.I.; Hinder, L.M.; Jackson, S.W.; Valesano, A.; Levinson, G.E.; Feldman, E.L. Dyslipidemia impairs mitochondrial trafficking and function in sensory neurons. FASEB J. 2018, 32, 195–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, E.P.; Coppey, L.J.; Calcutt, N.A.; Oltman, C.L.; Yorek, M.A. Diet.-induced obesity in sprague-dawley rats causes microvascular and neural dysfunction. Diabetes Metab. Res. Rev. 2010, 26, 306–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinder, L.M.; O’Brien, P.D.; Hayes, J.M.; Backus, C.; Solway, A.P.; Sims-Robinson, C.; Feldman, E.L. Dietary reversal of neuropathy in a murine model of prediabetes and metabolic syndrome. Dis. Model Mech. 2017, 10, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Fu, H.; Hou, J.F.; Jiao, K.; Costigan, M.; Chen, J. High energy diets-induced metabolic and prediabetic painful polyneuropathy in rats. PLoS ONE 2013, 8, e57427. [Google Scholar] [CrossRef]
- Kim, B.; Feldman, E.L. Insulin resistance in the nervous system. Trends Endocrinol. Metab. 2012, 23, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Coppey, L.J.; Shevalye, H.; Obrosov, A.; Davidson, E.P.; Yorek, M.A. Determination of peripheral neuropathy in high-fat diet fed low-dose streptozotocin-treated female C57Bl/6J mice and Sprague-Dawley rats. J. Diabetes Investig. 2018. [Google Scholar] [CrossRef] [Green Version]
- Lupachyk, S.; Watcho, P.; Hasanova, N.; Julius, U.; Obrosova, I.G. Triglyceride, nonesterified fatty acids, and prediabetic neuropathy: Role for oxidative-nitrosative stress. Free Radic. Biol. Med. 2012, 52, 1255–1263. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.; Coppey, L.J.; Davidson, E.P.; Yorek, M.A. Rat models of diet.-induced obesity and high fat/low dose streptozotocin type 2 diabetes: Effect of reversal of high fat diet compared to treatment with enalapril or menhaden oil on glucose utilization and neuropathic endpoints. J. Diabetes Res. 2015, 2015, 307285. [Google Scholar] [CrossRef] [Green Version]
- Groover, A.L.; Ryals, J.M.; Guilford, B.L.; Wilson, N.M.; Christianson, J.A.; Wright, D.E. Exercise-mediated improvements in painful neuropathy associated with prediabetes in mice. Pain 2013, 154, 2658–2667. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Chow | CAF | p Value |
|---|---|---|---|
| Blood Lipids (13 weeks) | |||
| Triglycerides (mg/dL) | 116.0 ± 7.3 | 157.0 ± 9.5 | 0.003 |
| HDL Cholesterol (mg/dL) | 58.23 ± 2.61 | 40.93 ± 1.80 | <0.001 |
| TyG Index | 9.87 ± 0.08 | 10.30 ± 0.08 | <0.001 |
| Terminal Data (15 weeks) | |||
| Body Weight (g) | 548 ± 11 | 765 ± 26 | <0.001 |
| Naso-anal Length (cm) | 26.7 ± 0.3 | 28.2 ± 0.2 | <0.001 |
| Circumference (cm) | 19.9 ± 0.2 | 22.8 ± 0.6 | <0.001 |
| Lee Index | 0.307 ± 0.002 | 0.324 ± 0.003 | <0.001 |
| Tibia Length (cm) | 4.4 ± 0.03 | 4.5 ± 0.03 | 0.003 |
| rpWAT (g) | 4.0 ± 0.4 | 14.3 ± 1.2 | <0.001 |
| Liver Weight (g) | 14.1 ± 0.4 | 18.9 ± 0.7 | <0.001 |
| Liver Score (0–3) | 0.36 ± 0.13 | 2.50 ± 0.17 | <0.001 |
| FBG (mmol/L) | 5.0 ± 0.1 | 5.5 ± 0.1 | 0.006 |
| Fasting Plasma Leptin (ng/mL) | 2.1 ± 0.4 | 5.7 ± 0.5 | <0.001 |
| Plasma IL-6 (pg/mL) | 891.0 ± 52.2 | 1173.1 ± 143.4 | 0.084 |
| Plasma TNF-α (pg/mL) | 1418 ± 115 | 2761 ± 563 | 0.036 |
| Parameters | Chow | CAF |
|---|---|---|
| Von-Frey 50% PWT (g) | 2.99 ± 0.39 | 3.98 ± 0.62 |
| Nerve Conduction Studies | ||
| Tail SNAP (μV) | 19.06 ± 1.73 | 19.91 ± 2.64 |
| Tail SNCV (m/s) | 34.01 ± 0.87 | 33.25 ± 0.84 |
| Tibial Amplitude (mV) | 7.49 ± 0.52 | 7.60 ± 0.64 |
| Sciatic MNCV (m/s) | 42.31 ± 1.59 | 40.43 ± 1.94 |
| Nerve Excitability Indices | ||
| TEd 10–20 ms (%) | 37.48 ± 0.81 | 36.01 ± 0.95 |
| Resting I/V (slope) | 1.04 ± 0.03 | 1.06 ± 0.04 |
| Refractoriness at 2.5 ms (%) | 6.20 ± 1.63 | 7.81 ± 1.24 |
| Superexcitability at 5 ms (%) | −0.65 ± 0.68 | 2.14 ± 0.83 * |
| Subexcitability (%) | 8.16 ± 0.69 | 8.00 ± 0.96 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.J.; Kendig, M.D.; Wild, B.M.; Issar, T.; Krishnan, A.V.; Morris, M.J.; Arnold, R. Evidence of Altered Peripheral Nerve Function in a Rodent Model of Diet-Induced Prediabetes. Biomedicines 2020, 8, 313. https://doi.org/10.3390/biomedicines8090313
Hossain MJ, Kendig MD, Wild BM, Issar T, Krishnan AV, Morris MJ, Arnold R. Evidence of Altered Peripheral Nerve Function in a Rodent Model of Diet-Induced Prediabetes. Biomedicines. 2020; 8(9):313. https://doi.org/10.3390/biomedicines8090313
Chicago/Turabian StyleHossain, Md Jakir, Michael D. Kendig, Brandon M. Wild, Tushar Issar, Arun V. Krishnan, Margaret J. Morris, and Ria Arnold. 2020. "Evidence of Altered Peripheral Nerve Function in a Rodent Model of Diet-Induced Prediabetes" Biomedicines 8, no. 9: 313. https://doi.org/10.3390/biomedicines8090313
APA StyleHossain, M. J., Kendig, M. D., Wild, B. M., Issar, T., Krishnan, A. V., Morris, M. J., & Arnold, R. (2020). Evidence of Altered Peripheral Nerve Function in a Rodent Model of Diet-Induced Prediabetes. Biomedicines, 8(9), 313. https://doi.org/10.3390/biomedicines8090313