
Capillary Electrophoresis–Mass Spectrometry with Multisegment Injection and In-Capillary Preconcentration for High-Throughput and Sensitive Determination of Therapeutic Decapeptide Triptorelin in Pharmaceutical and Biological Matrices

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Samples
2.2. Instrumentation
2.3. Capillary Treatment
2.4. Field-Enhanced Sample Injection (FESI) and Multisegment Injection (MSI)
2.5. Procedures for Sample and Standard Solution Preparation
3. Results and Discussion
3.1. Optimization of CZE Separation
3.2. Optimization of MS Detection
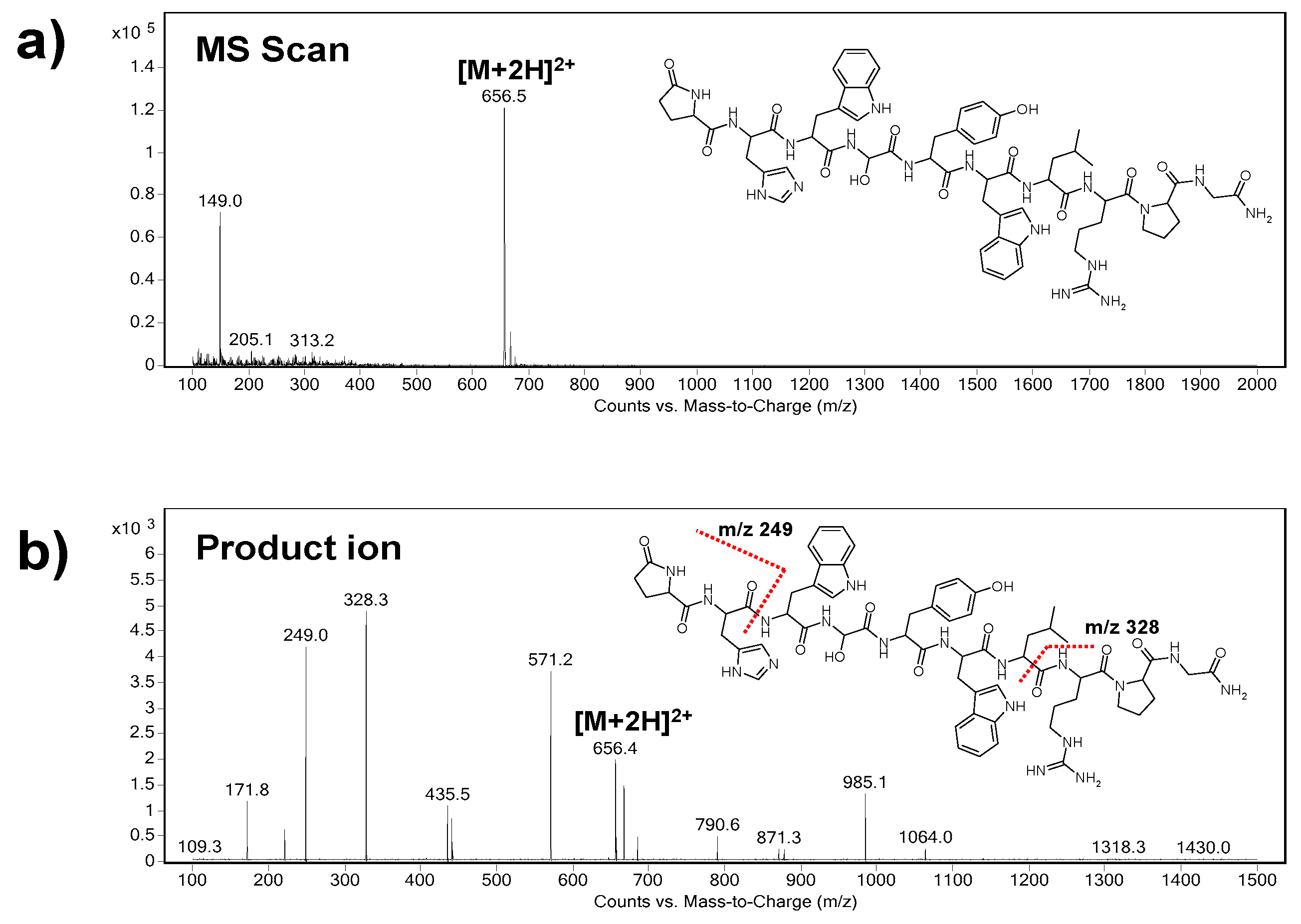
3.2.1. Electrospray Ionization (ESI) Step
3.2.2. MS/MS Step
3.3. In-Capillary Sample Preconcentration and Improvement of the Sample Throughput
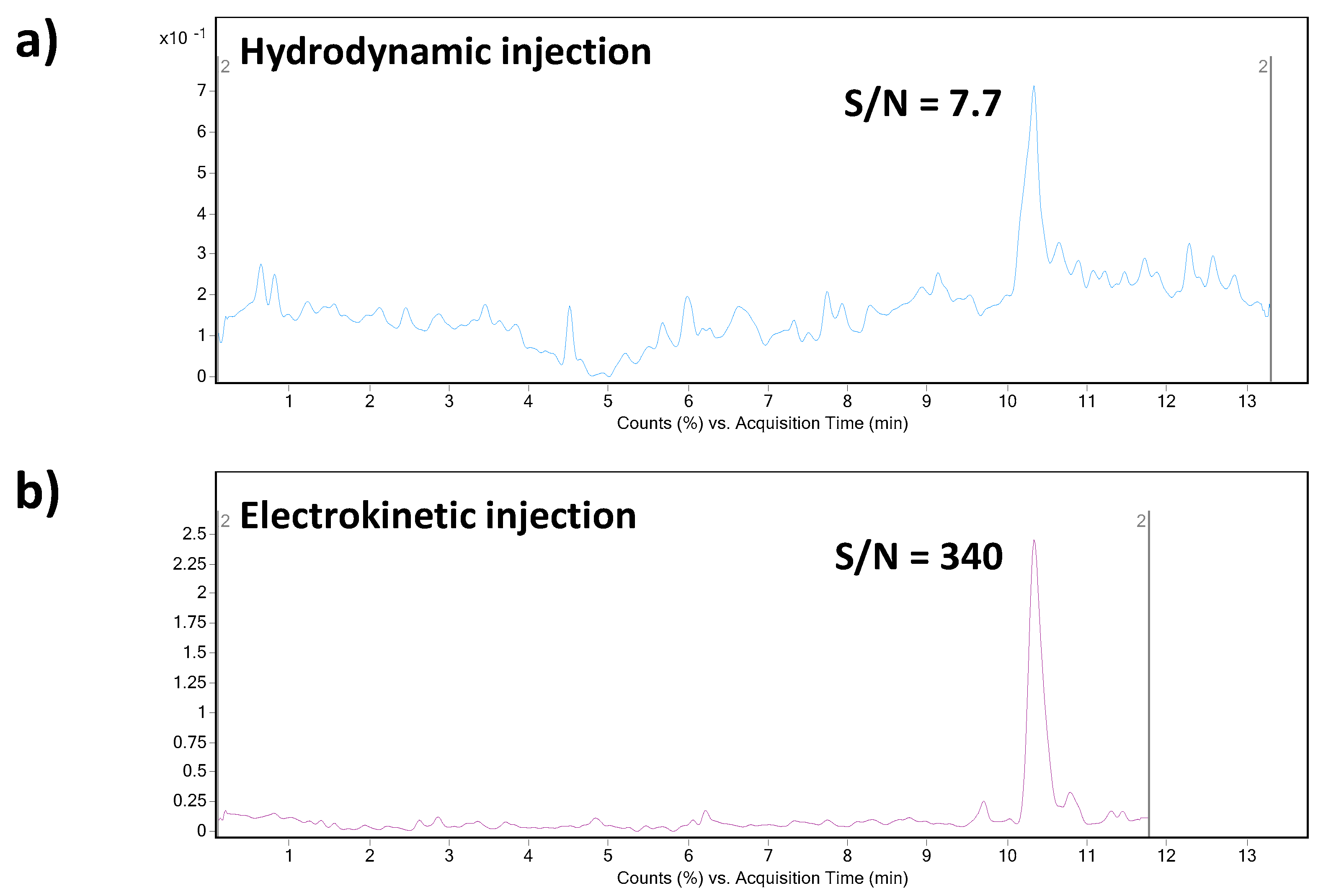
3.3.1. Field-Enhanced Sample Injection (FESI)
3.3.2. Multisegment Injection (MSI)
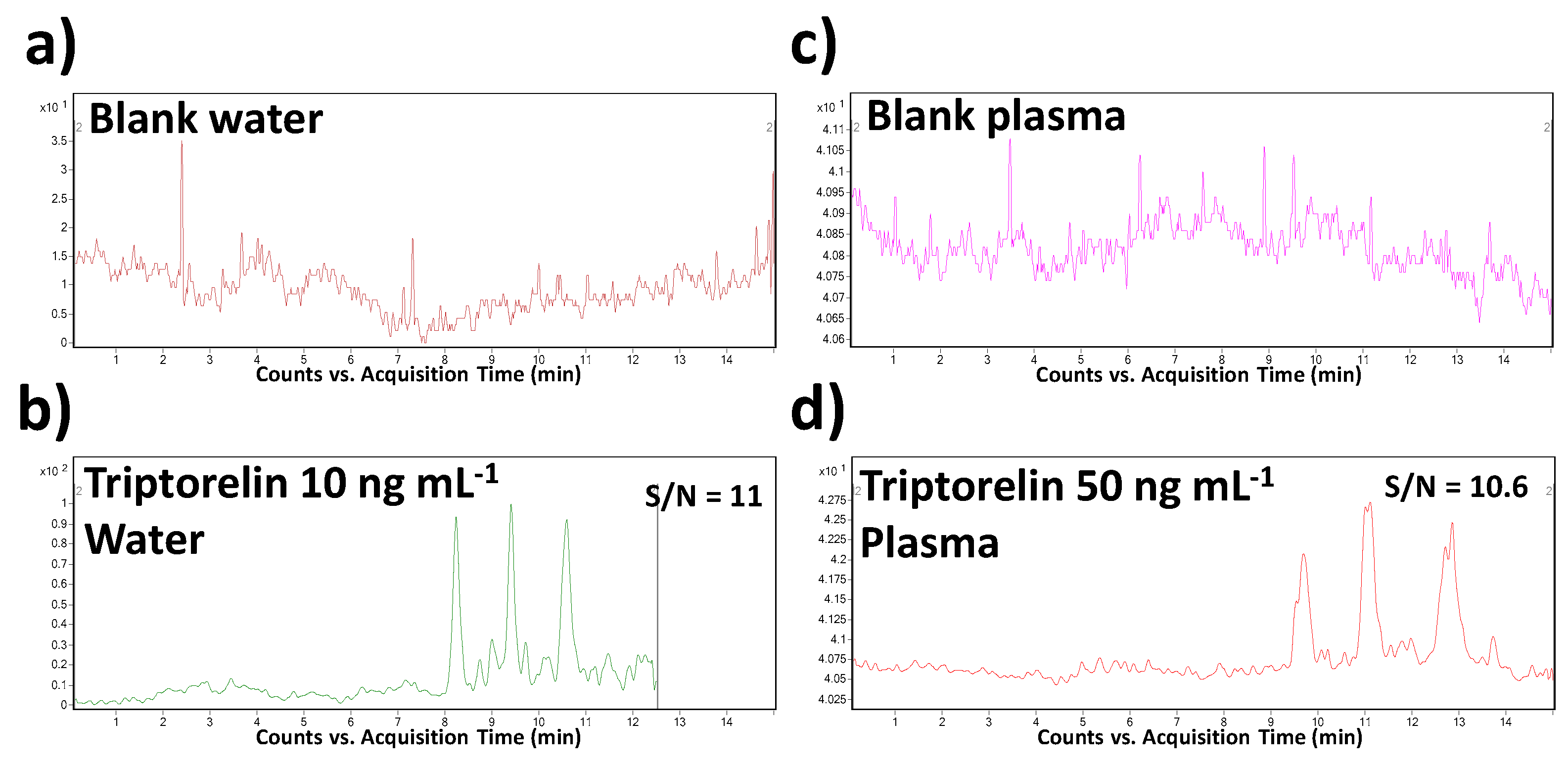
3.4. Method Validation
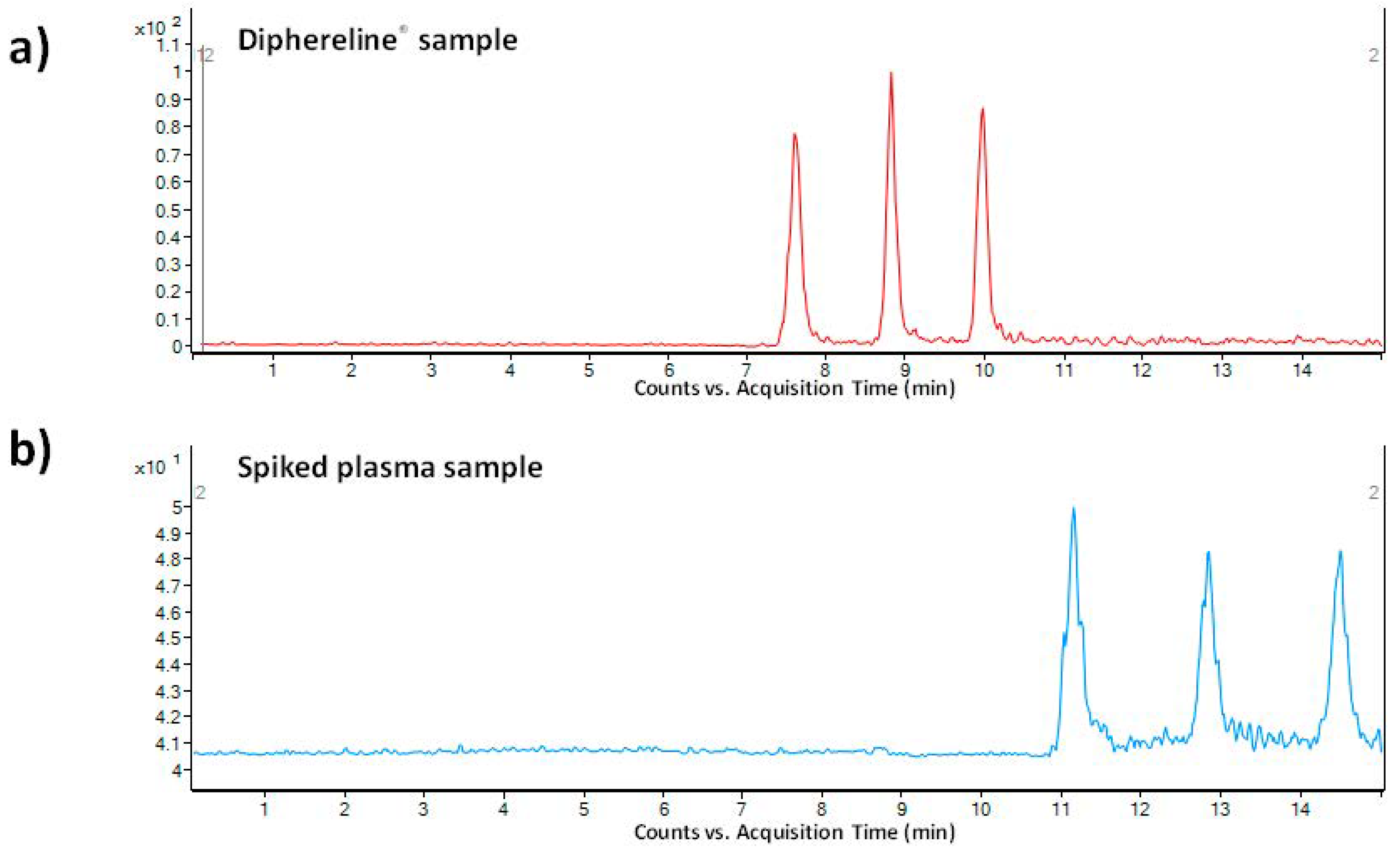
3.5. Method Application
3.6. Comparison of Methods for Triptorelin Analysis in Biological Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mahfouda, S.; Moore, J.K.; Siafarikas, A.; Zepf, F.D.; Lin, A. Puberty suppression in transgender children and adolescents. Lancet Diabetes Endo. 2017, 5, 816–826. [Google Scholar] [CrossRef]
- WADA. The 2020 Prohibited List. Available online: https://www.wada-ama.org/sites/default/files/wada_2020_english_prohibited_list_0.pdf (accessed on 19 November 2019).
- Li, W.; Zhang, J.; Tse, F.L.S. Handbook of LC-MS Bioanalysis, 1th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Van Andel, L.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Review of chromatographic bioanalytical assays for the quantitative determination of marine-derived drugs for cancer treatment. Mar. Drugs 2018, 16, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kočová Vlčková, H.; Pilařová, V.; Svobodová, P.; Plíšek, J.; Švec, F.; Nováková, L. Current stage of bioanalytical chromatography in clinical analysis. Analyst 2018, 143, 1305–1325. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.-A.; Xiang, X.; Ong, P.S.; Mitchell, E.Q.Y.; Syn, N.; Wee, I.; Kumar, A.P.; Yong, W.P.; Sethi, G.; Goh, B.C.; et al. A review on liquid chromatography-tandem mass spectrometry methods for rapid quantification of oncology drugs. Pharmaceutics 2018, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.H.; Bowden, P.; Du, Z.; Marshall, J.G. Mass spectrometry of peptides and proteins from human blood. Mass Spectrom. Rev. 2011, 30, 685–732. [Google Scholar] [CrossRef]
- Zemenova, J.; Sykora, D.; Adamkova, H.; Maletinska, L.; Elbert, T.; Marek, A.; Blechova, M. Novel approach to determine ghrelin analogs by liquid chromatography with mass spectrometry using a monolithic column. J. Sep. Sci. 2017, 40, 1032–1039. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, R.; Liu, S.; Yin, L.; Xu, T.; Fawcett, J.P.; Gu, J. Liquid chromatography tandem mass spectrometry with triple stage fragmentation for highly selective analysis and pharmacokinetics of alarelin in rat plasma. J. Sep. Sci. 2019, 42, 3033–3040. [Google Scholar] [CrossRef]
- Piestansky, J.; Barath, P.; Majerova, P.; Galba, J.; Mikus, P.; Kovacech, B.; Kovac, A. A simple and rapid LC-MS/MS and CE-MS/MS analytical strategy for the determination of therapeutic peptides in modern immunotherapeutics and biopharmaceutics. J. Pharm. Biomed. Anal. 2020, 189, 113449. [Google Scholar] [CrossRef]
- Stolz, A.; Jooß, K.; Höcker, O.; Römer, J.; Schlecht, J.; Neusüß, C. Recent advances in capillary electrophoresis-mass spectrometry: Instrumentation, methodology and applications. Electrophoresis 2019, 40, 79–112. [Google Scholar] [CrossRef] [Green Version]
- Voeten, R.L.C.; Ventouri, I.K.; Haselberg, R.; Somsen, G.W. Capillary electrophoresis: Trends and recent advances. Anal. Chem. 2018, 90, 1464–1481. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Nebot, V.; Benavente, F.; Toro, I.; Barbosa, J. Migration behaviour of therapeutic peptide hormones: Prediction of optimal separation by capillary electrophoresis. Electrophoresis 2001, 22, 4333–4340. [Google Scholar] [CrossRef]
- Sanz-Nebot, V.; Benavente, F.; Toro, I.; Barbosa, J. Evaluation of chromatographic versus electrophoretic behaviour of a series of therapeutic peptide hormones. J. Chromatogr. A 2003, 985, 411–423. [Google Scholar] [CrossRef]
- Sanz-Nebot, V.; Benavente, F.; Toro, I.; Barbosa, J. Capillary electrophoresis coupled to time of flight-mass spectrometry of therapeutic peptidehormones. Electrophoresis 2003, 24, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Nebot, V.; Benavente, F.; Toro, I.; Barbosa, J. Comparison of sheathless and sheath-flow electrospray interfaces for the capillary electrophoresis-electrospray ionization-mass spectrometry analysis of peptides. Electrophoresis 2005, 26, 1457–1465. [Google Scholar] [CrossRef]
- Progent, F.; Taverna, M.; Le Potier, I.; Gopée, F.; Ferrier, D. A study of the binding between polymers and peptides, using affinity capillary electrophoresis, applied to polymeric drug delivery systems. Electrophoresis 2002, 23, 938–944. [Google Scholar] [CrossRef]
- Pantůčková, P.; Gebauer, P.; Boček, P.; Křivánková, L. Electrolyte systems for on-line CE-MS: Detection requirements and separation possibilities. Electrophoresis 2009, 30, 203–214. [Google Scholar] [CrossRef]
- Ploch, M.; Bunz, S.-C.; Neusüß, C. Capillary electrophoresis/mass spectrometry relevant to pharmaceutical and biotechnological applications. Electrophoresis 2012, 33, 1517–1530. [Google Scholar]
- Herrero, M.; Ibañez, E.; Cifuentes, A. Capillary electrophoresis-electrospray mass spectrometry in peptide analysis and peptidomics. Electrophoresis 2008, 29, 2148–2160. [Google Scholar] [CrossRef] [Green Version]
- Scriba, G.K.E. Separation of peptides by capillary electrophoresis. In Capillary Electrophoresis. Methods in Molecular Biology, 1th ed.; Schmitt-Kopplin, P., Ed.; Humana Press: New York, NY, USA, 2016; pp. 365–391. [Google Scholar]
- Gomes, F.P.; Yates III, J.R. Recent trends of capillary electrophoresis-mass spectrometry in proteomics research. Mass Spectrom. Rev. 2019, 38, 445–460. [Google Scholar] [CrossRef]
- Klampf, C.K.; Himmelsbach, M. Sheath liquids in CE-MS: Role, parameters, and optimization. In Capillary Electrophoresis-Mass Spectrometry (CE-MS): Principles and Applications, 1th ed.; de Jong, G., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2016; pp. 41–63. [Google Scholar]
- Mazzarino, M.; Calvaresi, V.; de la Torre, X.; Parrotta, G.; Sebastianelli, C.; Botrè, F. Development and validation of a liquid chromatography-mass spectrometry procedure after solid-phase extraction for detection of 19 doping peptides in human urine. Forensic Toxicol. 2015, 33, 321–337. [Google Scholar] [CrossRef]
- Han, J.; Zhang, S.; Liu, W.; Leng, G.; Sun, K.; Li, Y.; Di, X. An analytical strategy to characterize the pharmacokinetics and pharmacodynamics of triptorelin in rats based on simultaneous LC-MS/MS analysis of triptorelin and endogenous testosterone in rat plasma. Anal. Bioanal. Chem. 2014, 406, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Kuehnbaum, N.L.; Kormendi, A.; Britz-McKibbin, P. Multisegment injection-capillary electrophoresis-mass spectrometry: A high-throughput platform for metabolomics with high data fidelity. Anal. Chem. 2013, 85, 10664–10669. [Google Scholar] [CrossRef] [PubMed]
- Kuehnbaum, N.L.; Gillen, J.B.; Kormendi, A.; Lam, K.P.; DiBattista, A.; Gibala, M.J.; Britz-McKibbin, P. Multiplexed separations for biomarker discovery in metabolomics: Elucidating adaptive responses to exercise training. Electrophoresis 2015, 36, 2226–2236. [Google Scholar] [CrossRef]
- Yamamoto, M.; Ly, R.; Gill, B.; Zhu, Y.; Moran-Mirabal, J.; Britz-McKibbin, P. Robust and High-Throughput Method for Anionic Metabolite Profiling: Preventing Polyimide Aminolysis and Capillary Breakages under Alkaline Conditions in Capillary Electrophoresis-Mass Spectrometry. Anal. Chem. 2016, 88, 10710–10719. [Google Scholar] [CrossRef]
- Saoi, M.; Kennedy, K.M.; Gohir, W.; Sloboda, D.M.; Britz-McKibbin, P. Placental metabolomics for assessment of sex-specific differences in fetal development during normal gestion. Sci. Rep. 2020, 10, 9399. [Google Scholar] [CrossRef]
- Shanmuganathan, M.; Kroezen, Z.; Gill, B.; Azab, S.; de Souza, R.J.; Teo, K.K.; Atkinson, S.; Subbaro, P.; Desai, D.; Anand, S.S.; et al. The maternal serum metabolome by multisegment injection-capillary electrophoresis-mass spectrometry: A high-throughput platform and standardized data workflow for large-scale epidemiological studies. Nat. Protoc. 2021, 16, 1966–1994. [Google Scholar] [CrossRef]
- Yamamoto, M.; Shanmuganathan, M.; Hart, L.; Pai, N.; Britz-McKibbin, P. Urinary metabolites enable differential diagnosis and therapeutic monitoring of pediatric inflammatory bowel disease. Metabolites 2021, 11, 245. [Google Scholar] [CrossRef]
- Igarashi, K.; Ota, S.; Kaneko, M.; Hirayama, A.; Enomoto, M.; Katumata, K.; Sugimoto, M.; Soga, T. High-throughput screening of salivary polyamine markers for discrimination of colorectal cancer by multisegment injection capillary electrophoresis tandem mass spectrometry. J. Chromatogr. A 2021, 1652, 462355. [Google Scholar] [CrossRef]
- Staub, A.; Rudaz, S.; Veuthey, J.-L.; Schappler, J. Multiple injection technique for the determination and quantitation of insulin formulations by capillary electrophoresis and time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 8041–8047. [Google Scholar] [CrossRef]
- Geiser, L.; Rudaz, S.; Veuthey, J.-L. Validation of capillary electrophoresis-mass spectrometry methods for the analysis of a pharmaceutical formulation. Electrophoresis 2003, 24, 3049–3056. [Google Scholar] [CrossRef]
- DiBattista, A.; Rampersaud, D.; Lee, H.; Kim, M.; Britz-McKibbin, P. High Throughput Screening Method for Systematic Surveillance of Drugs of Abuse by Multisegment Injection–Capillary Electrophoresis–Mass Spectrometry. Anal. Chem. 2017, 89, 11853–11861. [Google Scholar] [CrossRef]
- Boley, D.A.; Zhang, Z.; Dovichi, N.J. Multisegment injections improve peptide identification rates in capillary zone electrophoresis-based bottom-up proteomics. J. Chromatogr. A 2017, 1523, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Raedschelders, K.; Santos, M.; Van Eyk, J.E. Profiling B-Type Natriuretic Peptide Cleavage Peptidoforms in Human Plasma by Capillary Electrophoresis with Electrospray Ionization Mass Spectrometry. J. Proteome Res. 2017, 16, 4515–4522. [Google Scholar] [CrossRef]
- Saoi, M.; Sasaki, K.; Sagawa, H.; Abe, K.; Kogiso, T.; Tokushige, K.; Hashimoto, E.; Ohashi, Y.; Britz-McKibbin, P. High Throughput Screening of Serum γ-Glutamyl Dipeptides for Risk Assessment of Nonalcoholic Steatohepatitis with Impaired Glutathione Salvage Pathway. J. Proteome Res. 2020, 19, 2689–2699. [Google Scholar] [CrossRef]
- ICH Harmonised Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology. Step 4 Version. Euro-pean Medicines Agency, November 2005. Available online: http://www.ich.org/cache/compo/276-254-1.html (accessed on 18 July 2021).
- U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM). Bioanalytical Method Validation Guidance for Industry; U.S. FDA: Rockville, MD, USA, 2018.
- Minkov, N.K.; Zozikov, B.I.; Yaneva, Z.; Uldry, P.A. A phase II trial with new triptorelin sustained release formulations in prostatic carcinoma. Int. Urol. Nephrol. 2001, 33, 379–383. [Google Scholar] [CrossRef]
- Wang, J.; Kong, S.; Yan, J.; Jin, G.; Guo, Z.; Shen, A.; Xu, J.; Zhang, X.; Zou, L.; Liang, X. Hydrophilic interaction liquid chromatography-solid phase extraction directly combined with protein precipitation for the determination of triptorelin in plasma. J. Chromatogr. B 2014, 960, 214–221. [Google Scholar] [CrossRef]
- Han, J.; Sun, J.; Sha, C.; Zhang, J.; Gai, Y.; Li, Y.; Liu, W. Quantitation of slow release triptorelin in beagle dog plasma by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2012, 66, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.-H.; Lee, K.; Jeon, S.-H.; Song, S.H.; Yun, Y.-M.; Chun, S.; Kim, H.S.; Kim, J.Y.; In, M.K.; Song, J. Simultaneous measurement of serum chemical castration agents and testosterone levels using ultra-performance liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2016, 40, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, T.; Zhang, Z.; Fawcett, J.P.; Sun, D.; Gu, J. Micro-solid phase extraction and LC-MS(3) for the determination of triptorelin in rat plasma and application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2019, 166, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Görgens, C.; Guddat, S.; Thieme, D.; Dellanna, F.; Schänzer, W.; Thevis, M. Simplifying and expanding the screening for peptides <2 kDa by direct urine injection, liquid chromatography, and ion mobility mass spectrometry. J. Sep. Sci. 2016, 39, 333–341. [Google Scholar] [PubMed]
- Forsback, A.-P.; Noppari, P.; Viljanen, J.; Mikkola, J.; Jokinen, M.; Leino, L.; Bjerregaard, S.; Borglin, C.; Halliday, J. Sustained in-vivo release of triptorelin acetate from a biodegradable silica depot: Comparison to Pamorelin® LA. Nanomaterials 2021, 11, 1578. [Google Scholar] [CrossRef] [PubMed]
- Davarani, S.S.H.; Pourahadi, A.; Ghasemzadeh, P. Quantification of controlled release leuprolide and triptorelin in rabbit plasma using electromembrane extraction coupled with HPLC-UV. Electrophoresis 2019, 40, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Nojavan, S.; Bidarmanesh, T.; Mohammadi, A.; Yaripour, S. Electromembrane extraction of gonadotropin-releasing hormone agonists from plasma and wastewater samples. Electrophoresis 2016, 37, 826–833. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BGE | pH | tm (min) | RSDtm (%) | RSDarea (%) | N | S/N |
|---|---|---|---|---|---|---|
| 10 mM HFo | 2.91 | 9.47 | 1.8 | 6.4 | 14,441 | 33.3 |
| 20 mM HFo | 2.75 | 9.53 | 0.9 | 7.0 | 21,197 | 54.1 |
| 50 mM HFo | 2.54 | 9.66 | 2.5 | 6.2 | 20,041 | 55.8 |
| 50 mM HFo + 50 mM HAc | 2.85 | 10.42 | 0.5 | 20.7 | 24,552 | 52.5 |
| 1000 mM HFo | 1.88 | 10.53 | 1.3 | 4.6 | 27,683 | 52.6 |
| 10 mM NH4Fo + 20 mM HFo | 3.20 | 8.30 | 1.3 | 8.8 | 19,794 | 51.7 |
| Injection | RSDarea (%) | LOD (µg mL−1) | Reference |
|---|---|---|---|
| Hydrodynamical | 15 | 3.65 | [15] |
| Hydrodynamical | 33 | 2.25 | [16] |
| Hydrodynamical | 8.9 | 0.25 | This study |
| Electrokinetic | 5.5 | 0.005 | This study |
| Parameter | Water | Plasma |
|---|---|---|
| tm (min) | 10.53 | 14.54 |
| RSDtm (%), n = 6 | 0.24 | 0.99 |
| RSDarea (%), n = 6 | 7.61 | 11.01 |
| a (counts) | 27.98 | 97.88 |
| SDa, n = 6 | 1.12 | 11.99 |
| b (counts.ng mL−1) | 238.01 | 171.78 |
| SDb, n = 6 | 2.52 | 4.65 |
| r2 | 0.992 | 0.985 |
| LOD (ng.mL−1) | 5 | 25 |
| LOQ (ng.mL−1) | 10 | 50 |
| N | 36769 | 36956 |
| Nominal (µg mL−1) | Found (µg mL−1) | RSD (%) | RE (%) | ||||
|---|---|---|---|---|---|---|---|
| Matrix | Water | Plasma | Water | Plasma | Water | Plasma | |
| Intraday, n = 6 | 0.01 | 0.01 | - | 3.6 | - | −19.0 | - |
| 0.05 | 0.04 | 0.04 | 5.6 | 5.6 | −10.0 | −6.1 | |
| 0.1 | 0.11 | - | 9.4 | - | 9.0 | - | |
| 0.5 | 0.47 | - | 6.4 | - | −5.8 | - | |
| 1 | 0.95 | 0.93 | 2.5 | 4.8 | −4.6 | −7.5 | |
| 2 | 1.95 | - | 2.8 | - | −2.5 | - | |
| 5 | 5.24 | - | 5.5 | - | 4.8 | - | |
| 10 | 9.72 | 10.25 | 1.5 | 5.5 | −2.8 | 2.5 | |
| Interday, n = 6 | 0.01 | 0.01 | - | 4.2 | - | −19.8 | - |
| 0.05 | 0.04 | 0.04 | 4.4 | 15.4 | −14.2 | −6.1 | |
| 0.1 | 0.11 | - | 11.9 | - | 9.3 | - | |
| 0.5 | 0.50 | - | 8.8 | - | −0.1 | - | |
| 1 | 0.99 | 0.94 | 6.1 | 11.9 | −1.5 | −6.4 | |
| 2 | 1.93 | - | 4.0 | - | −3.5 | - | |
| 5 | 5.47 | - | 5.3 | - | 9.4 | - | |
| 10 | 9.61 | 9.66 | 2.3 | 11.4 | −3.9 | −3.4 | |
| Autosampler Stability | Freeze-to-Thaw Stability | |||||
|---|---|---|---|---|---|---|
| Low | Medium | High | Low | Medium | High | |
| Nominal (µg mL−1) | 0.05 | 1 | 10 | 0.05 | 1 | 10 |
| Found (µg mL−1) | 0.04 | 0.95 | 9.39 | 0.04 | 0.86 | 9.21 |
| Accuracy (RE %) | −18.2 | −4.8 | −6.1 | −19.4 | −13.9 | −7.9 |
| Method. | Study | Matrix | Sample preparation | t (min) | LOD (ng mL−1) | Ref. |
|---|---|---|---|---|---|---|
| LC–MS/MS | Pharmacokinetic study in rat | Plasma | Protein precipitation, solid phase extraction | ~14 | 6 | [42] |
| LC–MS/MS | Pharmacokinetic study in beagle dog | Plasma | Protein precipitation, solid phase extraction | ~14 | 0.006 | [43] |
| UHPLC–MS/MS | Monitoring of chemical castration | Serum | Protein precipitation, solid phase extraction | 12 | 0.25 | [44] |
| LC–MS/MS/MS | Pharmacokinetic study in rat | Plasma | Protein precipitation, micro-solid phase extraction | 5 | 0.006 | [45] |
| LC–MS/MS | Model study for antidoping | Urine | Centrifugation | 15 | 0.25 | [46] |
| LC–MS/MS | Pharmacokinetic study in rat | Plasma | Protein precipitation, solid phase extraction | 6 | 0.006 | [25] |
| LC–MS/MS | Pharmacokinetic study in rat | Plasma | Protein precipitation, solid phase extraction | 3 | 0.3 | [47] |
| LC–UV | Pharmacokinetic study in rabbit | Plasma | Dilution, electromembrane extraction | 12 | 0.15 | [48] |
| LC–UV | Model study of spiked human plasma | Plasma | Dilution, electromembrane extraction | 40 | 0.6 | [49] |
| CE–MS/MS | Model study of spiked human plasma | Plasma | Protein precipitation | 15 | 25 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piešťanský, J.; Čižmárová, I.; Štefánik, O.; Matušková, M.; Horniaková, A.; Majerová, P.; Mikuš, P. Capillary Electrophoresis–Mass Spectrometry with Multisegment Injection and In-Capillary Preconcentration for High-Throughput and Sensitive Determination of Therapeutic Decapeptide Triptorelin in Pharmaceutical and Biological Matrices. Biomedicines 2021, 9, 1488. https://doi.org/10.3390/biomedicines9101488
Piešťanský J, Čižmárová I, Štefánik O, Matušková M, Horniaková A, Majerová P, Mikuš P. Capillary Electrophoresis–Mass Spectrometry with Multisegment Injection and In-Capillary Preconcentration for High-Throughput and Sensitive Determination of Therapeutic Decapeptide Triptorelin in Pharmaceutical and Biological Matrices. Biomedicines. 2021; 9(10):1488. https://doi.org/10.3390/biomedicines9101488
Chicago/Turabian StylePiešťanský, Juraj, Ivana Čižmárová, Ondrej Štefánik, Michaela Matušková, Andrea Horniaková, Petra Majerová, and Peter Mikuš. 2021. "Capillary Electrophoresis–Mass Spectrometry with Multisegment Injection and In-Capillary Preconcentration for High-Throughput and Sensitive Determination of Therapeutic Decapeptide Triptorelin in Pharmaceutical and Biological Matrices" Biomedicines 9, no. 10: 1488. https://doi.org/10.3390/biomedicines9101488
APA StylePiešťanský, J., Čižmárová, I., Štefánik, O., Matušková, M., Horniaková, A., Majerová, P., & Mikuš, P. (2021). Capillary Electrophoresis–Mass Spectrometry with Multisegment Injection and In-Capillary Preconcentration for High-Throughput and Sensitive Determination of Therapeutic Decapeptide Triptorelin in Pharmaceutical and Biological Matrices. Biomedicines, 9(10), 1488. https://doi.org/10.3390/biomedicines9101488