
Fabrication of Strong Self-Reinforced Polyethylene Terephthalate Composites through the In Situ Nanofibrillation Technology



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
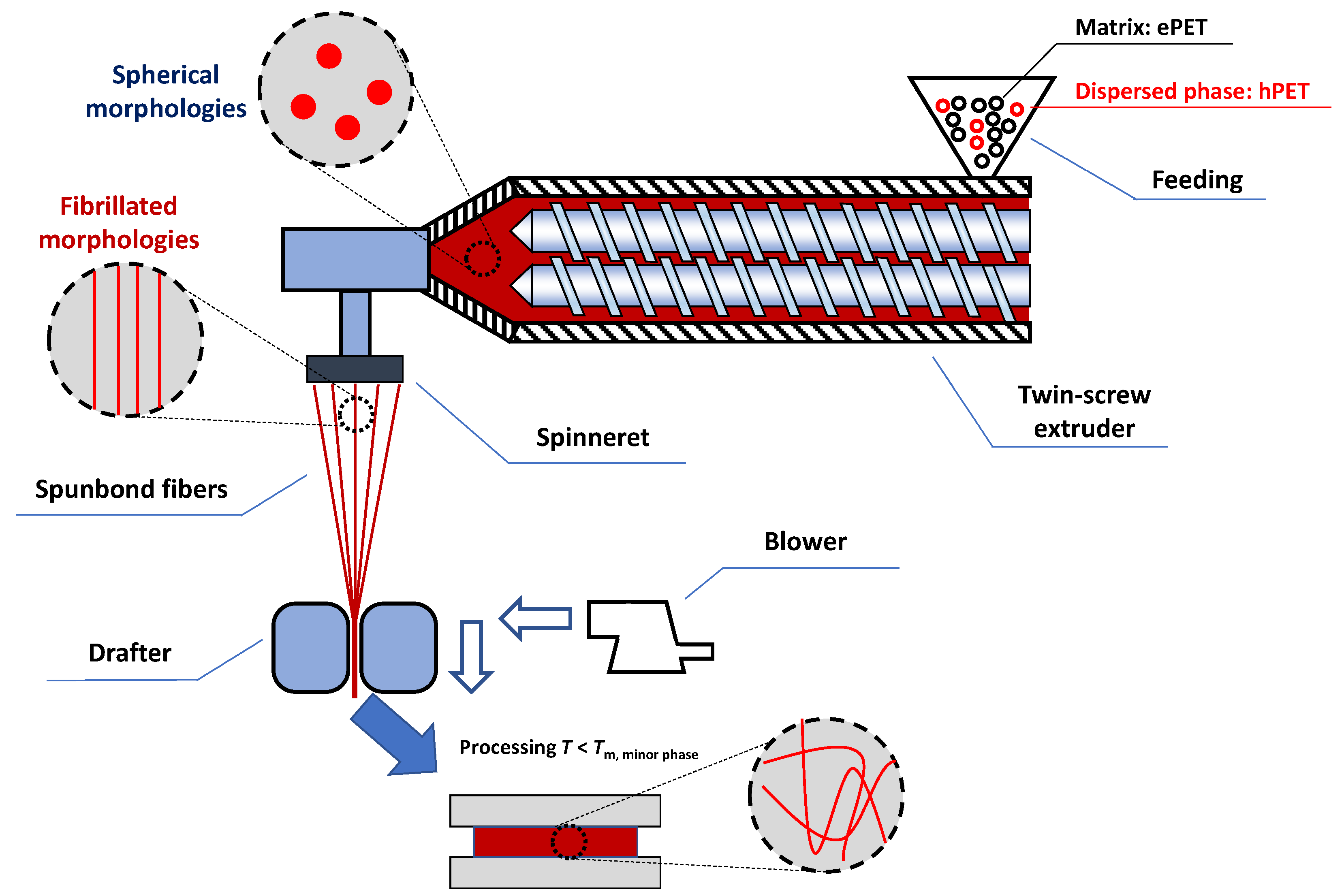
2.2. Polymer Blend Compounding and Spun Bond
2.3. Tensile Properties Measurement
2.4. Rheological Behavior Characterization
2.5. Foaming and Characterization
2.6. Characterization of Thermal Conductivity
3. Results and Discussion
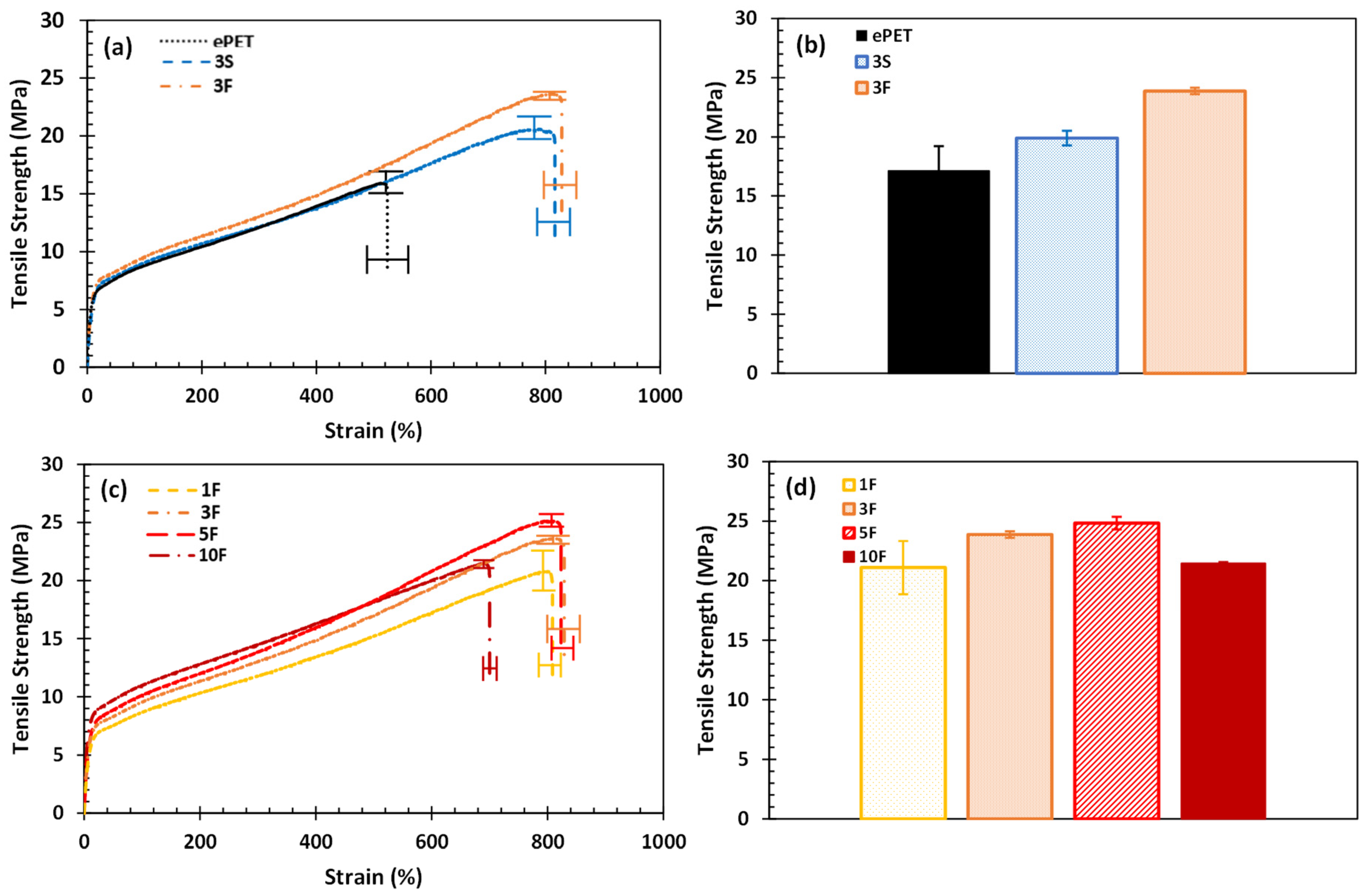
3.1. Tensile Properties of Composites
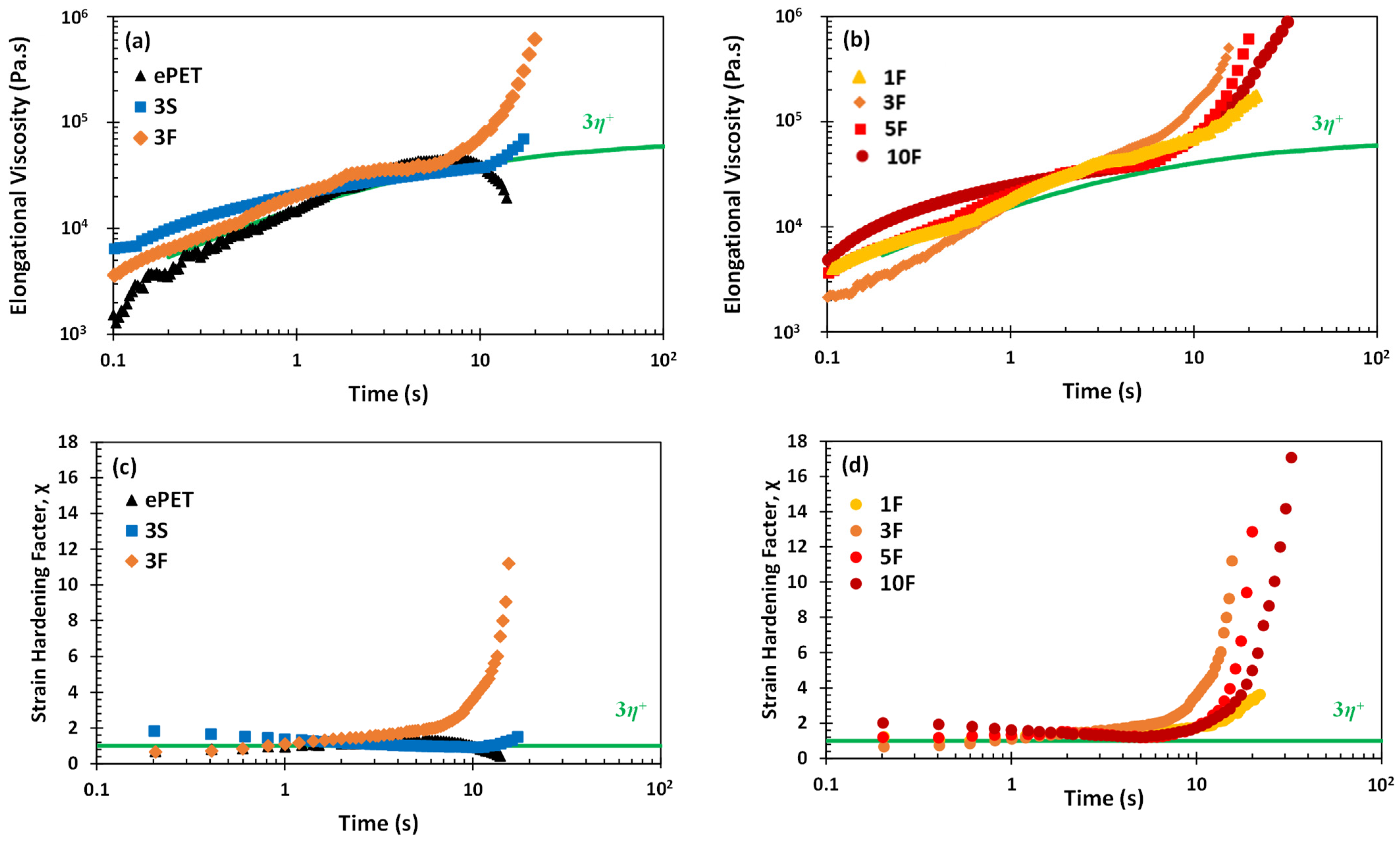
3.2. Rheological Properties of Composites
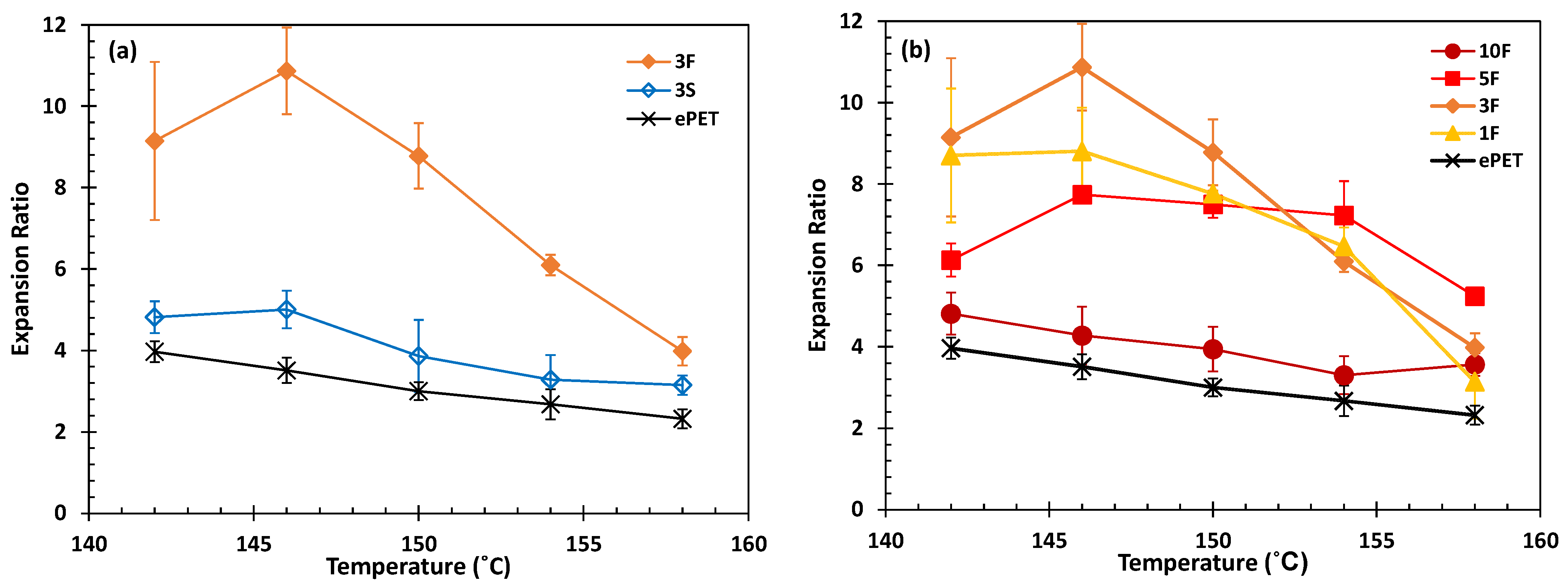
3.3. Foaming and Foam Characterization
3.4. Thermal Conductivity of Foams
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Walther, G.; Post, E.; Convey, P.; Menzel, A.; Parmesan, C.; Beebee, T.J.C.; Fromentin, J.; Hoegh-Guldberg, O.; Bairlein, F. Ecological response to recent climate change. Nature 2002, 416, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Wu, C.M.; Pu, F.C.; Chiu, C.H. Fabrication and mechanical properties of self-reinforced poly(ethylene terephthalate) composites. Express Polym. Lett. 2011, 5, 228–237. [Google Scholar] [CrossRef]
- Peijs, T. Composites for recyclability. Mater. Today 2003, 6, 30–35. [Google Scholar] [CrossRef]
- Matabola, K.P.; De Vries, A.R.; Moolman, F.S.; Luyt, A.S. Single polymer composites: A review. J. Mater. Sci. 2009, 44, 6213–6222. [Google Scholar] [CrossRef]
- Kmetty, Á.; Bárány, T.; Karger-Kocsis, J. Self-reinforced polymeric materials: A review. Prog. Polym. Sci. 2010, 35, 1288–1310. [Google Scholar] [CrossRef]
- Fakirov, S.; Duhovic, M.; Maitrot, P.; Bhattacharyya, D. From PET nanofibrils to nanofibrillar single-polymer composites. Macromol. Mater. Eng. 2010, 295, 515–518. [Google Scholar] [CrossRef]
- Capiati, N.J.; Porter, R.S. The concept of one polymer composites modelled with high density polyethylene. J. Mater. Sci. 1975, 10, 1671–1677. [Google Scholar] [CrossRef]
- Hine, P.J.; Ward, I.M.; Jordan, N.D.; Olley, R.H.; Bassett, D.C. A comparison of the hot-compaction behavior of oriented, high-modulus, polyethylene fibers and tapes. J. Macromol. Sci. Part B 2001, 40, 959–989. [Google Scholar] [CrossRef]
- Hees, T.; Zhong, F.; Koplin, C.; Jaeger, R.; Mulhaupt, R. Wear resistant all-PE single-component composites via 1D nanostructure formation during melt processing. Polymer 2018, 151, 47–55. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Kuang, X.; Wang, J.; Wang, D.; Li, L.; Yan, S. A scanning electron microscopy study on the morphologies of isotactic polypropylene induced by its own fibers. Macromolecules 2004, 37, 2847–2853. [Google Scholar] [CrossRef]
- Barany, T.; Karger-Kocsis, J.; Czigany, T. Development and characterization of self-reinforced poly(propylene) composites: Carded mat reinforcement. Polym. Adv. Technol. 2006, 17, 818–824. [Google Scholar] [CrossRef]
- Wnag, J.; Song, F.; Ding, Y.; Shao, M. The incorporation of graphene to enhance mechanical properties of polypropylene self-reinforced polymer composites. Mater. Des. 2020, 195, 1090733. [Google Scholar]
- Jiang, J.; Liu, X.; Lian, M.; Pan, Y.; Chen, Q.; Liu, H.; Zheng, G.; Guo, Z.; Schubert, D.W.; Shen, C.; et al. Self-reinforcing and toughening isotactic polypropylene via melt sequential injection molding. Polym. Test. 2018, 67, 183–189. [Google Scholar] [CrossRef]
- Majola, A.; Vainionpää, S.; Rokkanen, P.; Mikkola, H.M.; Törmälä, P. Absorbable self-reinforced polylactide (SR-PLA) composite rods for fracture fixation: Strength and strength retention in the bone and subcutaneous tissue of rabbits. J. Mater. Sci. Mater. Med. 1992, 3, 43–47. [Google Scholar] [CrossRef]
- Törmälä, P. Biodegradable self-reinforced composite materials; Manufacturing structure and mechanical properties. Clin. Mater. 1992, 10, 29–34. [Google Scholar] [CrossRef]
- Zhang, J.M.; Reynolds, C.T.; Peijs, T. All-poly(ethylene terephthalate) composites by film stacking of oriented tapes. Compos. Part A Appl. Sci. Manuf. 2009, 40, 1747–1755. [Google Scholar] [CrossRef]
- Andrejewski, J.; Przyszczypkowski, P.; Szostak, M. Development and characterization of poly(ethylene terephthalate) based injection molded self-reinforced composites. Direct reinforcement by overmolding the composite inserts. Mater. Des. 2018, 153, 273–286. [Google Scholar] [CrossRef]
- Morye, S.S.; Hine, P.J.; Duckett, R.A.; Carr, D.J.; Ward, I.M. Comparison of the properties of hot compacted gel-spun polyethylene fibre composites with conventional gel-spun polyethylene fibre composites. Compos. Part A Appl. Sci. Manuf. 1999, 30, 649–660. [Google Scholar] [CrossRef]
- Tohidi, S.D.; Rocha, A.M.; Dencheva, N.V.; Denchev, Z. Single polymer laminate composites by compression molding of knitted textiles and microparticles of polyamide 6: Preparation and structure-properties relationship. Compos. Part A Appl. Sci. Manuf. 2018, 109, 171–183. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, E.S.; Romero-Diez, S.; Wang, Y.; Huang, G.; Li, A.; Yang, Y.; Lee, P.C. Cyclic olefin copolymer foam: A promising thermal insulation material. Chem. Eng. J. 2021, 409, 128251. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, E.S.; Romero-Diez, S.; Wang, Y.; Huang, G.; Li, K.; Yang, Y.; Lee, P.C. Supercritical fluid foaming of nanoscale phase patterned structures: An approach to lightweight hierarchical porous foams with superior thermal insulation. Chem. Eng. J. 2022, 431, 133490. [Google Scholar] [CrossRef]
- Rizvi, A.; Andalib, Z.K.M.; Park, C.B. Fiber-spun polypropylene/polyethylene terephthalate microfibrillar composites with enhanced tensile and rheological properties and foaming ability. Polymer 2017, 110, 139–148. [Google Scholar] [CrossRef]
- Evstatiev, M.; Fakirov, S. Microfibrillar reinforcement of polymer blends. Polymer 1992, 33, 877–880. [Google Scholar] [CrossRef]
- Hsissou, R.; Bekhta, A.; Dagdag, O.; El Bachiri, A.; Rafik, M.; Elharfi, A. Rheological properties of composite polymers and hybrid nanocomposites. Heliyon 2020, 6, e04187. [Google Scholar] [CrossRef]
- Rahman, S.S.; Mahmud, M.B.; Monfared, A.R.; Lee, P.C.; Park, C.B. Achieving outstanding toughness of PMMA while retaining its strength, stiffness, and transparency using in situ developed TPEE nanofibrils. Compos. Sci. Technol. 2023, 236, 109994. [Google Scholar] [CrossRef]
- Zhai, W.; Wang, H.; Yu, J.; Dong, J.; He, J. Cell coalescence suppressed by crosslinking structure in polypropylene microcellular foaming. Polym. Eng. Sci. 2008, 48, 1312–1321. [Google Scholar] [CrossRef]
- Stange, J.; Munstedt, H. Effect of long-chain branching on the foaming of polypropylene with azodicarbonamide. J. Cell. Plast. 2006, 42, 445–467. [Google Scholar] [CrossRef]
- Zhai, W.; Park, C.B. Effect of nanoclay addition on the foaming behavior of linear polypropylene-based soft thermoplastic polyolefin foam blown in continuous extrusion. Polym. Eng. Sci. 2011, 51, 2387–2397. [Google Scholar] [CrossRef]
- Guo, G.; Wang, K.H.; Park, C.B.; Kim, Y.S.; Li, G. Effects of nanoparticles on the density reduction and cell morphology of extruded metallocene polyethylene/wood fiber nanocomposites. J. Appl. Polym. Sci. 2007, 104, 1058–1063. [Google Scholar] [CrossRef]
- Kim, E.S.; Park, H.E.; Lopez-Barron, C.R.; Lee, P.C. Enhanced foamability with shrinking microfibers in linear polymer. Polymers 2019, 11, 211. [Google Scholar] [CrossRef]
- Jalali, A.; Romero-Diez, S.; Nofar, M.; Park, C.B. Entirely environment-friendly polylactide composites with outstanding heat resistance and superior mechanical performance fabricated by spunbond technology: Exploring the role of nanofibrillated stereocomplex polylactide crystals. Int. J. Biol. Macromol. 2021, 193, 2210–2220. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Son, S.; Chun, S.; Kang, T.J.; Kwak, S. Nonisothermal Crystallization Behavior of Exfoliated Poly (ethylene terephthalate) -Layered Silicate Nanocomposites in the Presence and Absence of Organic Modifier. J. Polym. Sci. 2008, 46, 989–999. [Google Scholar] [CrossRef]
- Li, P.; Lan, B.; Zhang, Q.; Yang, Q.; Gong, P.; Park, C.B. Microcellular foams simultaneous reinforcing and toughening strategy of combining nano-fibrillation network and supercritical solid-state foaming. Polymer 2022, 252, 124928. [Google Scholar] [CrossRef]
- Almanza, O.; Rodríguez-Pérez, M.A.; De Saja, J.A. Applicability of the transient plane source method to measure the thermal conductivity of low-density polyethylene foams. J. Polym. Sci. Part B Polym. Phys. 2004, 2, 1226–1234. [Google Scholar] [CrossRef]
- He, Y.; Jia, S.; Fang, C.; Tan, L.; Qin, S.; Yin, X.; Park, C.B.; Qu, J. Constructing synergistically strengthening-toughening 3D network bundle structures by stereocomplex crystals for manufacturing high-performance thermoplastic polyurethane nanofibers reinforced poly(lactic acid) composites. Compos. Sci. Technol. 2023, 232, 109847. [Google Scholar] [CrossRef]
- Zeidi, M.; Kim, C.I.; Park, C.B. The role of interface on the toughening and failure mechanisms of thermoplastic nanocomposites reinforced with nanofibrillated rubber. Nanoscale 2021, 13, 20248–20280. [Google Scholar] [CrossRef]
- Embabi, M.; Kweon, M.S.; Anstey, A.; Tuccito, A.V.; Park, C.B.; Shivokhin, M.E.; Lee, P.C. Effect of Ethylene Comonomer Content on the Foam Processing Window of Long-Chain Branched Polypropylene. ACS Appl. Polym. Mater. 2023, 5, 2417–2429. [Google Scholar] [CrossRef]
- Embabi, M.; Kweon, M.S.; Wang, Y.X.; Lin, T.P.; Lopez-Barron, C.R.; Lee, P.C. Foaming Performance of Linear Polypropylene Ionomers. Macromolecules 2022, 55, 5645–5655. [Google Scholar] [CrossRef]
- Hong, J.S.; Ahn, K.H.; Lee, S.J. Strain hardening behavior of polymer blends with fibril morphology. Rheol. Acta 2005, 45, 202–208. [Google Scholar] [CrossRef]
- Wagner, M.H.; Yamaguchi, M.; Takahashi, M. Quantitative assessment of strain hardening of low-density polyethylene melts by the molecular stress function model. J. Rheol. 2003, 47, 779–793. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Ianniruberto, G.; Greco, F.; Marrucci, G. Primitive chain network simulations for branched polymers. Rheol. Acta 2006, 46, 297–303. [Google Scholar] [CrossRef]
- Leung, S.N.; Wong, A.; Park, C.B.; Zong, J.H. Ideal surface geometries of nucleating agents to enhance cell nucleation in polymeric foaming processes. J. Appl. Polym. Sci. 2008, 108, 3997–4003. [Google Scholar] [CrossRef]
- Anstey, A.; Chang, E.; Kim, E.S.; Rizvi, A.; Kakroodi, A.R.; Park, C.B.; Lee, P.C. Nanofibrillated polymer systems: Design, application, and current state of the art. Prog. Polym. Sci. 2021, 113, 101346. [Google Scholar] [CrossRef]
- Guo, Y.; Lyu, Z.; Yang, X.; Lu, Y.; Ruan, K.; Wu, Y.; Kong, J.; Gu, J. Enhanced thermal conductivities and decreased thermal resistances of functionalized boron nitride/polyimide composites. Compos. Part B Eng. 2019, 164, 732–739. [Google Scholar] [CrossRef]
- Kuhn, J.; Ebert, H.P.; Arduini-Schuster, M.C.; Büttner, D.; Fricke, J. Thermal transport in polystyrene and polyurethane foam insulations. Int. J. Heat Mass Transf. 1992, 35, 1795–1801. [Google Scholar] [CrossRef]
- Gong, P.; Wang, G.; Tran, M.; Buahom, P.; Zhai, S.; Li, G.; Park, C.B. Advanced bimodal polystyrene/multi-walled carbon nanotube nanocomposite foams for thermal insulation. Carbon 2017, 120, 1–10. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, Q.; Wang, C.; Guo, B.; Park, C.B.; Wang, G. High thermal insulation and compressive strength polypropylene foams fabricated by high-pressure foam injection molding and mold opening of nano-fibrillar composites. Mater. Des. 2017, 131, 1–11. [Google Scholar] [CrossRef]
- Forest, C.; Chaumont, P.; Cassagnau, P.; Swoboda, B.; Sonntag, P. Polymer nano-foams for insulating applications prepared from CO2 foaming. Prog. Polym. Sci. 2015, 41, 122–145. [Google Scholar] [CrossRef]
- Erukhimovich, I.; de la Cruz, M.O. Phase equilibrium and charge fractionation in polyelectrolyte solutions. J. Polym. Sci. Part B Polym Phys. 2007, 45, 3003–3009. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Wang, W.; Gong, P.; Yang, Q.; Park, C.B.; Li, G. Ultra-fast degradable PBAT/PBS foams of high performance in compression and thermal insulation made from environment-friendly supercritical foaming. J. Supercrit. Fluids 2022, 181, 105512. [Google Scholar] [CrossRef]
- Hasanadeh, R.; Azdast, T.; Lee, P.C.; Park, C.B. A review of the state-of-the-art on thermal insulation performance of polymeric foams. Therm. Sci. Eng. Prog. 2023, 41, 101808. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
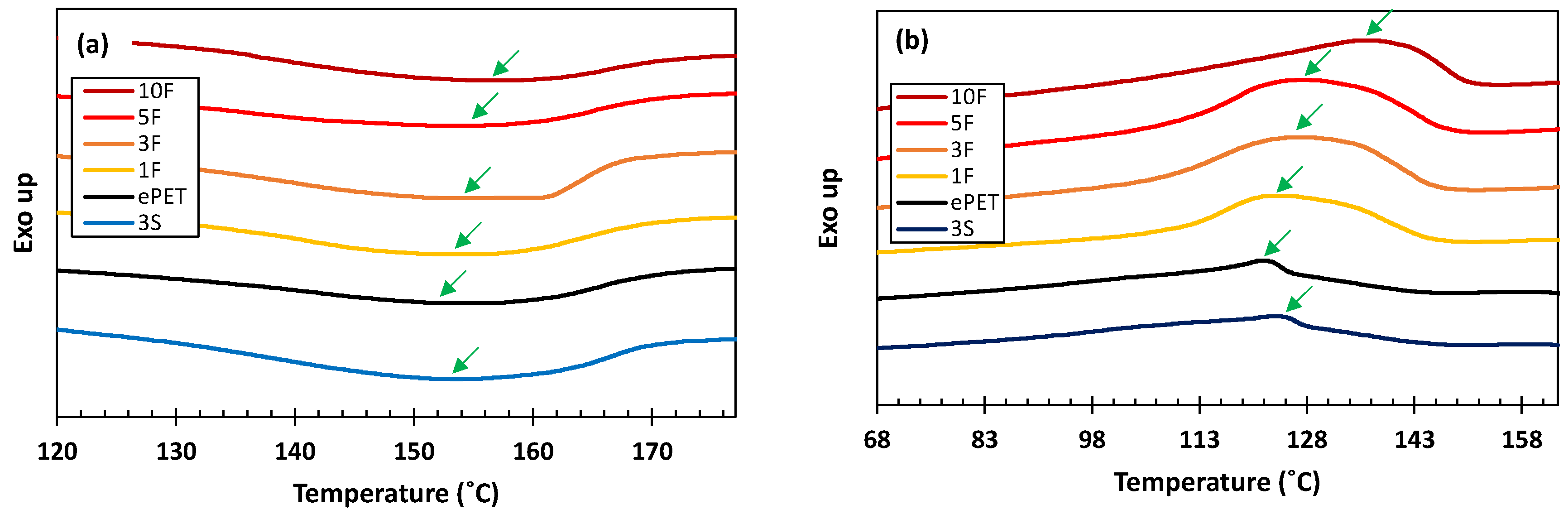
| Sample Name | Morphology of Minor Phase | Matrix ePET Content (wt%) | Minor Phase hPET Content (wt%) | Tm (°C) | Tc (°C) | (J/g) | (%) |
|---|---|---|---|---|---|---|---|
| ePET | 100 | 0 | 152.0 | 121.7 | 8.12 | 6.91 | |
| 1S | Spherical | 99 | 1 | 152.8 | 122.7 | 8.54 | 7.34 |
| 3S | 97 | 3 | 153.6 | 123.5 | 8.93 | 7.83 | |
| 5S | 95 | 5 | 154.1 | 124.1 | 9.11 | 8.15 | |
| 10S | 90 | 10 | 154.4 | 124.6 | 9.25 | 8.74 | |
| 1F | Fibrillated | 99 | 1 | 153.9 | 124.1 | 12.70 | 10.91 |
| 3F | 97 | 3 | 154.4 | 126.6 | 14.32 | 12.55 | |
| 5F | 95 | 5 | 154.9 | 127.0 | 13.46 | 12.05 | |
| 10F | 90 | 10 | 156.8 | 136.2 | 10.79 | 10.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.S.; Lee, P.C. Fabrication of Strong Self-Reinforced Polyethylene Terephthalate Composites through the In Situ Nanofibrillation Technology. Processes 2023, 11, 1434. https://doi.org/10.3390/pr11051434
Kim ES, Lee PC. Fabrication of Strong Self-Reinforced Polyethylene Terephthalate Composites through the In Situ Nanofibrillation Technology. Processes. 2023; 11(5):1434. https://doi.org/10.3390/pr11051434
Chicago/Turabian StyleKim, Eric S., and Patrick C. Lee. 2023. "Fabrication of Strong Self-Reinforced Polyethylene Terephthalate Composites through the In Situ Nanofibrillation Technology" Processes 11, no. 5: 1434. https://doi.org/10.3390/pr11051434
APA StyleKim, E. S., & Lee, P. C. (2023). Fabrication of Strong Self-Reinforced Polyethylene Terephthalate Composites through the In Situ Nanofibrillation Technology. Processes, 11(5), 1434. https://doi.org/10.3390/pr11051434