1. Introduction
The sheer diversity of genomic and phenotypic variation of CHO cell populations (and of other immortalized cell lines) is poorly understood and possibly underestimated. Also, mechanisms resulting in mutational evolution of populations in cell culture in general have not been appreciated. This is surprising, since numerous aspects of this diversity, as seen on the basis of chromosome structures and karyotypes, were established already five to six decades ago. Disturbing as these remarks may be, reliable and high quality pharmaceuticals from such cells have been given to hundreds of thousands of patients without any adverse effects. Resulting from dialogs between the industry and regulatory agencies steps and procedures had been established, since the start of using CHO cells for pharmaceutical production, to reduce any perceived or real risks related to emergence of mutations or other genetic changes in protein generating cell populations. While the main focus of this article will be a summary and discussion of facts and mechanisms behind rapid evolution and mutational change of CHO populations, some of the critical steps for the establishment of reliable and stable CHO processes will be discussed in the latter part of these lines.
2. Cell Line, Process Stability, and Cloning
Cell line and process stability are important for the manufacturing of pharmaceutical biologics. Assurance of a high quality product over years is the most important principle for the manufacturing of biopharmaceuticals. In pointing out contributing parameters to quality and consistency, recent communications by health product regulating authorities have demanded “proof of clonality” of recombinant CHO cell populations. The well-intended but incorrect regulatory suggestion is that cell line and process stability are linked to clonality. Most likely this is understood in a sense that the singularity of a cell and the derivation of dividing daughter cells from such a single cell would provide a (more) robust genetic foundation for reliable manufacture of recombinant proteins using cells. To the first author of this article, it seems that an emphasis on proof of clonality is being more strongly imposed by regulators today than this was discussed in the past. Having been involved hands-on in the generation of the first CHO derived products (Activase
® tPA, Pulmozyme
® DNase, etc.), the proof term was not used in the 1980s and 1990s. At the time, cloning was done in the first authors’ laboratory with cloning rings and cotton swabs from adherent cell cultures and many cells from a colony of cells were transferred into the next larger vessel for cell line establishment and testing. While the visual inspection seemed to imply that cells of an attached colony were in fact derived from a single cell, this did not constitute a proof of clonality and other more relevant tests were executed later in the process of cell line and manufacturing process development to assure the quality and consistency of proteins derived from such recombinant cells. Co-authored by scientists of leading US-based biopharmaceutical companies (with help from European based contributors, including the first author of this article) a ‘White Paper’, addressing the same topic, has been published recently. This publication emphasizes product quality testing and other process related steps as more important and more relevant approaches to the issue in question [
1].
We aim to expand on the White Paper to add facts and insights on cloning of CHO cells. We will consider for this discussion Darwinian (evolution) trends that are affecting the genetics of such immortalized cells in a profound manner. Our notes will also, so it is hoped, provide a path forward to further improve quality and consistency of products derived from recombinant CHO or other cell lines.
3. A Short Review on the Early History of CHO Cells
CHO cells have a culture history of more than 50 years, since they were established from the ovary of a Chinese Hamster and became “immortal” in the laboratory of Dr. Theodore Puck in the late 1950s [
2,
3]. For the sake of simplicity, we call the first immortalized cells ‘CHO-ori’—other names will be discussed later. CHO-ori cells were quickly becoming popular for the study of mammalian genetics and mammalian cell physiology due to two factors:
- (a)
The diploid hamster genome consists of only 11 pairs of easily identifiable and large chromosomes and thus modifications in their structures could be observed under a light microscope with great ease. Cells derived from the hamster showed these large chromosomes as well, although with significant structural modifications of the karyotypes. Thus some intentional or explored physiological changes of these cells were investigated for mappable hallmarks on chromosomes.
- (b)
The cell populations were robust and grew fast. Induced or spontaneous mutagenesis resulted, and could be appreciated when appropriate screening techniques were applied, in the establishment of clonal sublines with desired metabolic traits [
3].
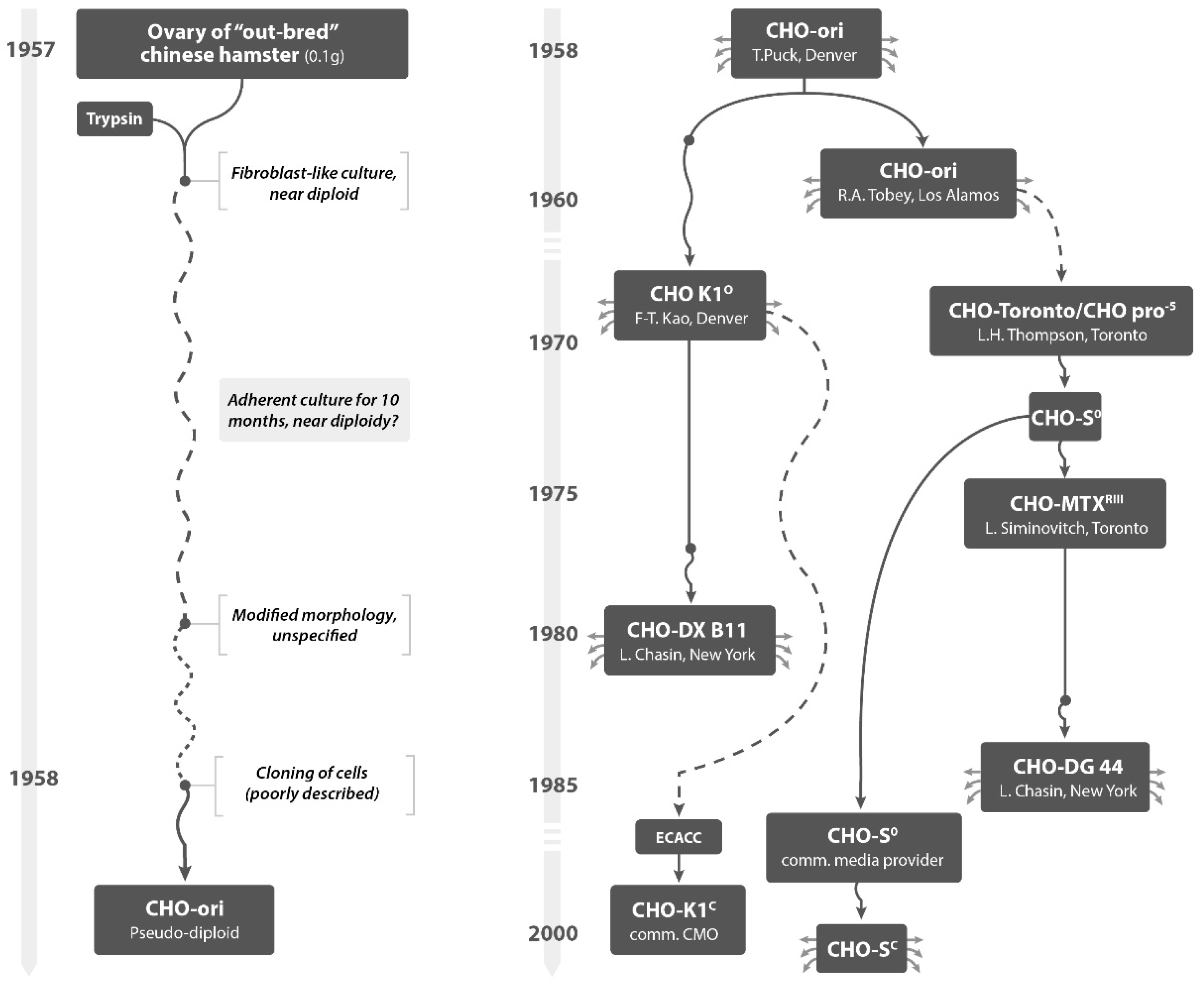
CHO cell populations (CHO-ori and others,
Figure 1) were handled by a number of labs and passed to many researchers in the field—many under names that are not popular anymore. However, the following names are still in use today: K1, DXB11, S, DG 44 (always with the prefix CHO): High-value protein therapeutics are made with derived cell populations, referring sometimes to these names. While some of the available literature provides locations and names of researchers where cells were handled [
4], the precise tracking of progeny of differently-named CHO cell lines cannot be done. Also and worse, establishing the number of subcultivations with different media and different culture systems from the early CHO-ori cultures until today is impossible. We do not know how many replication cycles occurred before any of these cell populations ended up in a laboratory for commercial manufacture and in a Master Cell Bank. National institutions for cell and microbial collections (ATCC, ECACC, or similar) will not provide a reconstructed culture history. Hundreds of such subcultivations occurred before a cell line, recombinant or not, was characterized genetically or tested in any way before entering into manufacture of pharmaceuticals. Each of the cell populations, obtained under a given name from a third party, has been exposed to a diverse range of culture conditions, some including the use of animal products. Most of these (named) cell lines have now been adapted to suspension culture and/or were liberated from growth in media that included the use of bovine or other sera. This has become a standard for the protein producing industry today.
All industry used names for CHO cells are based on the poorly recorded history of these cells, for which the first author of this paper attempted to generate a pedigree, based on an intensive study of the available literature. There are no tests available that certifies the naming and character of a given host cell population handled in one laboratory from another host cell line handled under a different name in another laboratory. Genomic sequencing allows today to verify the Hamster Origin of all these cells, but no publication has established a verifiable difference, for example, between CHO-K1 derived cell lines used today and cell lines that are traced back, by name, to the cell line established in the 1970s in Thompson’s laboratory as CHO-S. All cell lines with names dating back to the 1960s to 1990s of the last century are in the public domain.
4. Chromosomal and Genomic Heterogeneity of CHO Cells, Other Cell Lines, and Cancer Cells
Analyzing CHO cell lines in the 1960s and 1970s showed that their karyotypes, i.e., the structure and composition of the chromosomes, change frequently and apparently without an eventual stabilization. Such changes could be identified by simply looking at Giemsa stained metaphase spreads of chromosomes of as few of 20–30 cells [
5,
6,
7,
8]. These studies included the finding that mutations occur in CHO cells at a rate of 100 in 10
6 cells over a 30-day period of culture [
9]. Very recent studies on cells of non-diploid origins, including genome sequence analysis both with immortalized cells and with patient derived cancer cells emphasized their enormous fluidity and mutational impacts [
10,
11,
12,
13]. Chromosomes of such cells are subject to frequent and poorly understood events that rearrange, fracture or “pulverize” them and/or multiply individual chromosomal fragments and chromosomes. These events can partially or completely impair the structure of the majority of the original chromosomes as they are derived from the diploid organism. Concomitantly, a trend towards homozygosity is found in such cells. HeLa cells have an overall homozygosity status of 23%, more than 40% of the genome was found to be triploid, and some chromosomal fragments appear to be present in five and higher copy numbers—all homozygous in sequence [
10]. Interestingly, the overall genome size of immortalized cells, in the number of nucleotides, seems to stay relatively close to the diploid status of the animal organism.
DNA sequencing “averages” the nucleotide sequences found in a sample. This does not matter much with diploid genomes, since the sequence variation between allelic chromosomes are small and mutational variation between individual cells in an organism is small as well. The same occurs for karyotyping—of the 100 metaphases usually analyzed, only very few (1–3%) differ from a “perfect“ diploid status—i.e., the data reveal a perfect chromosome set, as established for a given animal or plant species. In immortalized cells there is an entirely different situation: Karyotype analysis and genome sequencing provide data on an averaged status of cells in a population that differ from cell to cell in chromosome structure and DNA sequence to a very large extent. In the karyotype analysis of CHO-K1 cells in 1973 [
7] only 9 of the typical 22 chromosomes of the hamster could be matched in cultivated cells on average. Remaining chromosomes showed drastically rearranged structures and its numbers varied from 19 to 24 in individual cells. In addition, a small number of cells showed a pseudo-tetraploid chromosome set of >35 chromosomes. Thus, the cell-to-cell variation in genetics, visible even with the low-resolution of a Giemsa-banded karyotype points to a significant problem in defining ‘a’ genome of CHO cells.
The publication of genome sequences of a CHO K1-derived cell population in 2011 [
14,
15] represents such an averaged sequence of a cell population sequenced more than 40 years after the reported cloning of these cells! Besides the time difference, the diversity of genomic structures in cells is not accessible by sequencing, since single cell sequencing has not been done so far, to our knowledge. Thus, the reader of this or any other genomic sequencing paper on CHO cells is faced with a serious dilemma regarding the interpretation and use of such data—particularly if such data are to guide efforts to generate new recombinant cell lines with modified genetic and physiological features. Even the perceived absence of a given sequence—possibly coding for an important enzyme—cannot be assured with absolute certainty, if a chromosomal fragment containing the gene in question is present in a percentage within the population of cells at a frequency too low to be detected within the averaged genome sequence. Another problem in interpreting and understanding the “averaged” genome sequences of CHO cells is the fact that a chromosome-map based sequence of the reference animal
Cricetulus griseus (as has been done for many other species) is not still available today.
Cloning has
not been found to reduce or stabilize chromosomal structure heterogeneity. To the contrary: A study done in our laboratory showed as much chromosomal heterogeneity of cloned cell lines after transfection, as before found in the karyotype(s) of the host cell line for transfection [
16]. Karyotyping of the clonally derived cell lines was done only a few weeks after their establishment. While the relatedness of the clonally derived cell lines (16 in total) to the parental non-transfected cell host could be identified to a certain degree, mostly with the help of the five to seven non-modified Hamster chromosomes, other chromosomes showed additional modifications of the karyotype not observed before.
A recent paper presented images of single karyotypes derived from metaphase spreads prepared with cells of five CHO host cell lines (two K1-derived, two DUXB11- and one DG44-derived cell population) [
17]. The authors found these cell lines showing a large diversity of genotypes (pseudo-diploid, pseudo triploid, mixed pseudo diploid and triploid) with highly variable chromosomal structures. While the karyotyping results are not entirely conclusive and probably insufficient to make strong statements on common chromosomal structures between the different cell lines, the largest and most visible “constant” in those karyotypes—as determined by Giemsa banding—was Chromosome 1 (the largest chromosome) of the Hamster. Other identifiable chromosomes of the diploid Hamster genome may be recognizable with a more detailed analysis of metaphase spreads, but apparently the majority of Hamster chromosomes were found only as single structures (no apparent homologs found).
5. How Fast Are Genomic and Other Mutations Emerging in CHO Cells?
How frequent are these mutations in a cell population, independently from steps that are considered as experimental cloning? To assume that the genomic structure and content of the emergent cell population is identical to the original clonally derived cell is incorrect. While ‘genomic relatedness’ is assured, the degree of relatedness to that unique first cell is variable and highly unpredictable. Genomic diversity in populations will depend on the rate of mutations and, possibly over time, the emergence of a balanced genotype and its’ corresponding phenotype most fit for the conditions in which the cells reside. Balance, and thus replicative stability of a given set of divergent genotypes is likely to evolve when environmental modifications or changes in the culture of these cells are kept small, even upon scale up. Evolutionary principles do apply at all phases of scale up cultures.
Emergence and stabilization of divergent, yet genetically related genotypes in animals, plants, and microorganisms can result in the separation of a species from another. Such species separations are driven by selective forces, including inherent mutational rates and environmental factors like changes of climate, isolation, emergence of new parasites, etc. Stabilization and maintenance of a separating genotype between two species can usually be maintained as long as the environmental conditions remain stable and cross breeding between the separated species populations is prevented. In CHO cells, the species-defining gene-pool sharing does not occur—i.e., any degree of relatedness from a given single cell genome (the “clone”) will depend on the selective environment in which these cells are propagated. It is also reasonable to assume that more extended cultures will give rise to genotypes more distant (different) from the genotype of the single cell.
The rate of mutations in immortalized cells is high. Data from several sources indicate that diversity in genomic structure can emerge in days and, if one looks carefully, in hours and minutes, as discussed in the following:
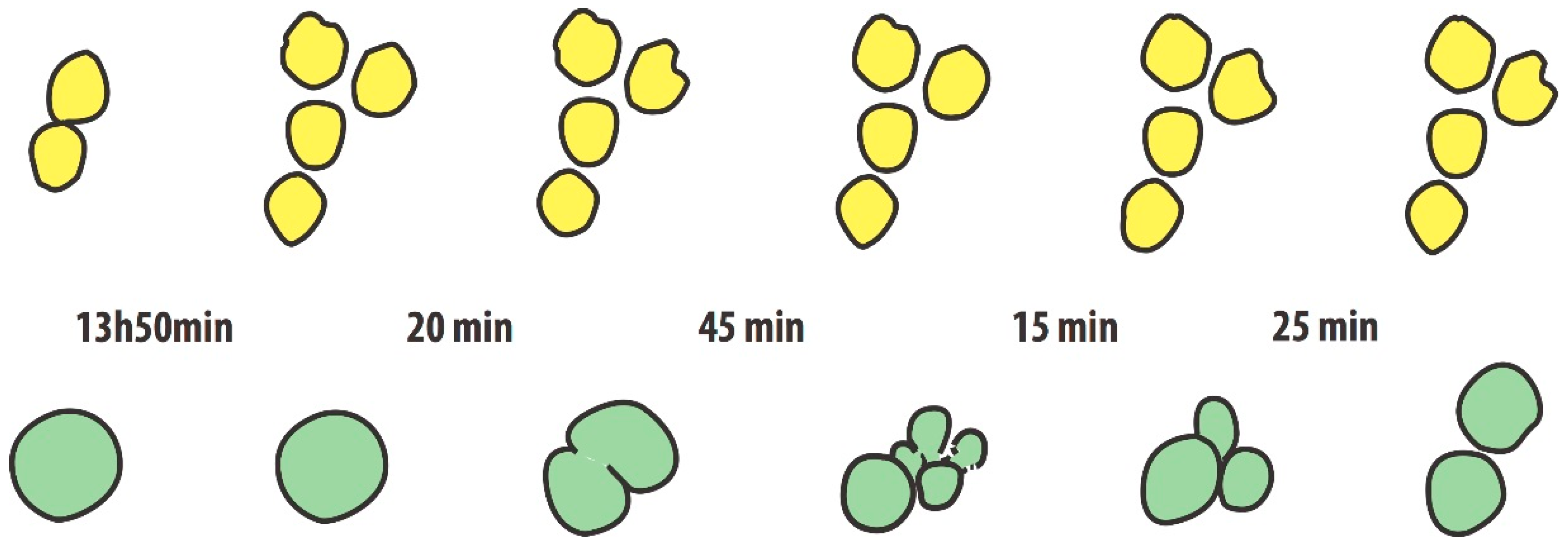
During his doctoral thesis, M. Jordan analyzed light-microscopic video-images of non-shaken hybridoma cultures. He could trace the fate of individual cells over several replication cycles [
18]. One of the striking observations was the following: Two cells derived from a prior cell division (i.e., clonal sisters), presented significant differences in time accrued until the next cell division. One daughter cell divided after 13 h, the other only after 20 h. Another set of images (
Figure 2), taken over a period of 18 h, shows three cells in the first image resting on the surface of the culture chamber. One of the cells is larger in diameter than typical, whereas the two others had just divided (they touch each other). About 14 h later, the two cells have divided again to generate a set of four daughter cells, whereas the ‘big one’ still sits untouched. However, another 20 min later, the big cell “divides“ over a period of 2 h into two and then five cells (or cell fragments) to then fuse to three and then to two cells. The figure shows a hand-drawn image of these events, derived from the original black and white photographic images. While such divisions and fusions could be the cause for tetraploidization of cells we really do not know what drives these phenomena that are considered ‘unusual’. However, they should not to be taken as rare. While hybridoma cultures may be considered a case of immortalized cells with a high propensity for chromosomal instability (due to the enforced fusion of mouse cells for their generation), anybody who has looked into CHO cultures by microscopy will verify the rather frequent occurrence of cells that are significantly larger in diameter than majority of cells (1–5%). Big cells are a strong indicator of a large nucleus, containing more chromosomes than typical. Thus the presence of cells whose genome has been enlarged due to entire or partial duplications of their chromosome set appear not to be a rare event in CHO cells.
The second example of rapid and frequent emergence of genetically diverse structures in clonal or non-clonally derived cell populations comes from studies on gene-amplification using methotrexate (MTX) or methionine sulphoximine (MSX). The first authors’ work on MTX treated CHO cells contributed to the understanding of these phenomena [
19]. In methotrexate treated CHO cells, massive genome restructuring can be selected for in days. Typical procedures, still applied widely today, ask for two to three weeks’ exposure to such selective environments, resulting in populations of cells that show a large diversity of rearranged chromosomal structures [
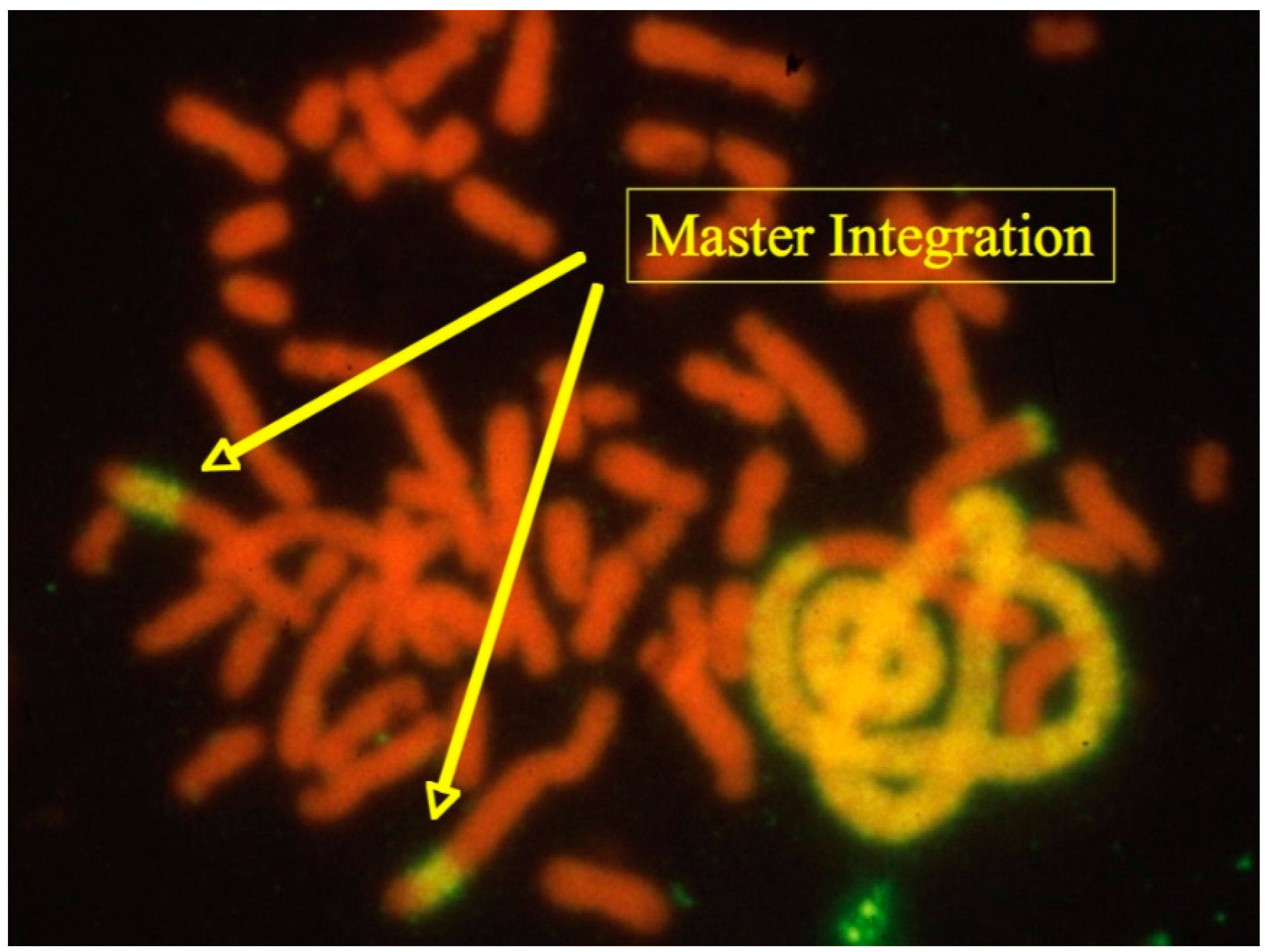
20]. One striking example of these is shown in
Figure 3 exhibiting several highly amplified chromosomal fragments containing DHFR sequences and the gene of interest in a single pseudo-tetraploid cell (>40 chromosomes).
The two “Master Integrations” shown in
Figure 3 are presenting each about 200 copies of linked DHFR and gene of interest DNA sequences within a large metacentric CHO chromosome. These particular chromosomes also show non-hybridizing ends, an indication for functional telomeric structures. Based on their overall structure (ratio of lengths in comparison to other chromosomes, centromere position, position of the hybridizing signals), Master Integrations could be identified in the majority of cells as a single copy in cultures that have been maintained in the absence of methotrexate (on which copy-number assessments were done). Reasonably, it was assumed that this chromosome provides the genetic stability of the genes of interest within the population of cells. The striking hybridization signals linked to one or two chromosomes towards the right of the image indicates the past occurrence of multiple catastrophic cell division scenarios (genome duplication, chromosomal breaks and fusions): If this cell would have gone through another mitosis, the structures of these highly elongated chromosomes containing amplified DNA would not have been faithfully replicated (because of lack of telomers) and thus daughter cells with divergent genotypes result.
The observations made on “gene amplified” CHO chromosomes are in general agreement with a large body of scientific studies by karyotyping and other techniques from the 1980s/1990s, on methotrexate treated cultured cells and cells derived from MTX-treated patients. Obviously, while any rearrangement of chromosomal DNA occurs spontaneously and are typically rare, the increase of the number of cells in a population with this rearranged DNA to a level to be detectable is the result of selection, favoring such cells in the environment in which they are cultured. The observed chromosomal rearrangements, seen under drug selection in a large majority of cells (30–60%) are frequently unstable structures. In CHO cells, many of these structures become depleted from the culture within a week or so when the selective agent is removed [
21]. However, some of them appear to be relatively stable entities within the population (such as the Master Integration described above) and are contributing to the elevated level of productivity seen for the product of interest. All rearranged structures are assumed to consist of large repetitive segments of DHFR plasmid derived DNA linked to the DNA of interest (encoding the protein therapeutic). The plasmid sequences are embedded into CHO genomic DNA, co-amplified together with the integrated DNA.
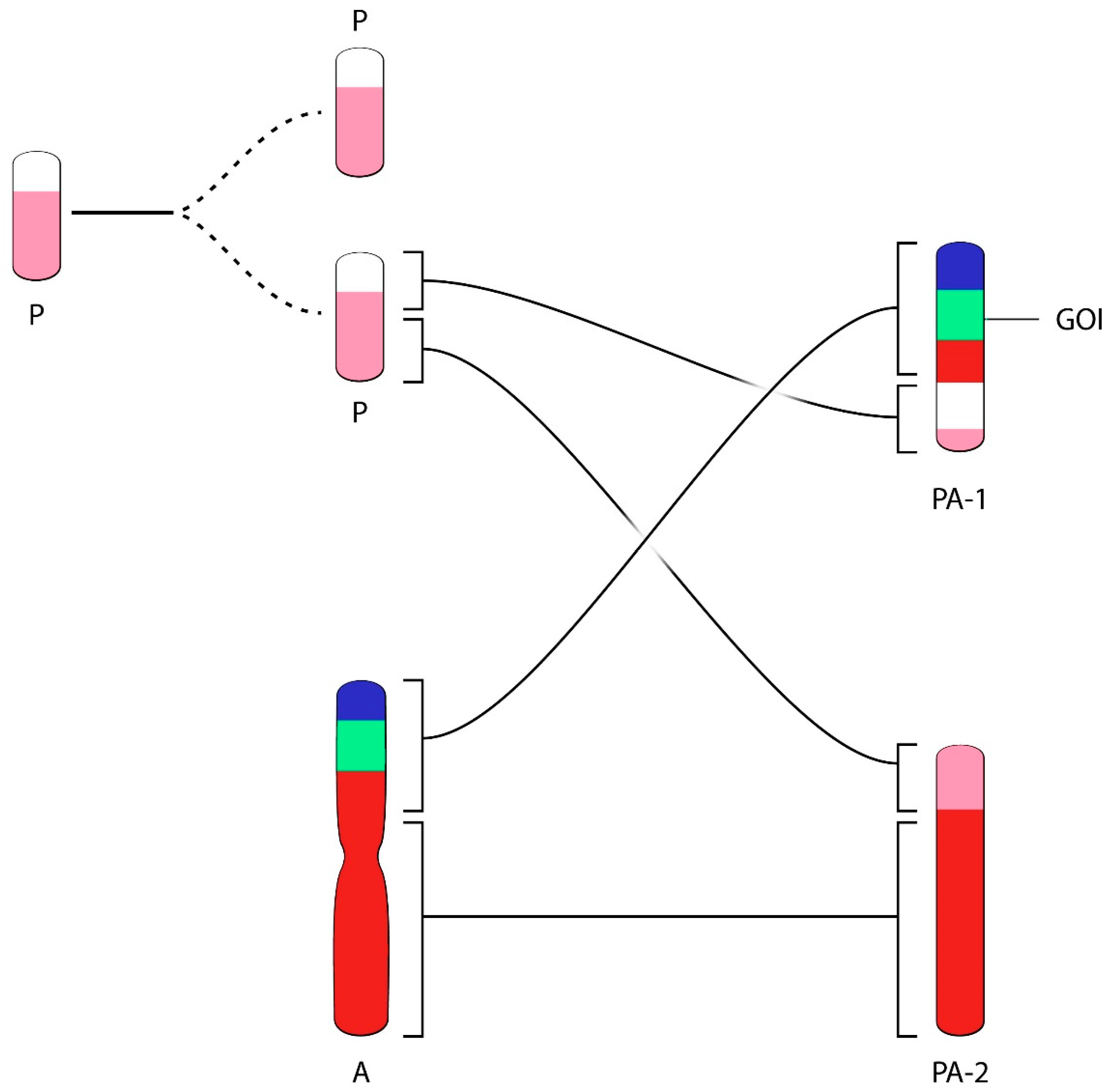
A third example of rapid rearrangement of chromosome structures containing transgenic DNA in a CHO producer cell line is provided in the excellent paper of the Merck group [
22]. Broly and co-workers used fluorescence in Situ hybridization (FISH) and chromosome painting technologies in their work. This method allows recognizing each CHO chromosome or chromosome fragment on the basis of specific color-labels of the original Hamster chromosomes. The producer cell line showed transgenic (and gene-amplified) DNA in the largest visible chromosome (Chromosome A), a composite of Chromosome 1 (red) and Chromosome 4 (blue) of the Hamster (
Figure 4). When studying cells of an additional Working Cell Bank, derived from a validated Master Cell Bank, a different chromosome bearing the genes of interest was detected upon extended culture of cells derived from the WCB. Cells from the new Working Cell Bank did not show any unusual structures. The WCB cells were intended as a starting point for the generation of a product marketed since many years. However, FISH analysis of cells from this new cell bank, studied at the level of a Post Production Cell Bank (PPCB) or an Extended Cell Bank (ExCB), showed an unusual position of the transgenic DNA in 12–15% of metaphases prepared from these cells. Other PPCBs or ExCB showed lower percentages of this unusual chromosome structure. Apparently, the unusual chromosome structures found in a low percentage of cells did not affect the product yield and product quality (personal communication). In studying all chromosome-based images one concludes that the population of cells used for production had derived from one unique and single cell (i.e., the population for production was of clonal origin). The transgenic DNA (green in
Figure 4, GOI) in chromosome A (red-green-blue) was definable as a unique genetic marker for the majority of cells analyzed. For clarity, we modified the label of chromosomes in the paper according to geneticists’ norm: Original Hamster Chromosomes are labeled by numbers and ordered by size 1, 2… etc. Chromosomes of CHO cells were labeled A, B, C, etc. also ordered by size. None of the chromosomes of the production cell line appear to contain a non-rearranged or otherwise non-modified structure. This chromosome was likely present in the vast majority of cells during most of the early phases of development of the cell line, the MCB generation and probably in the large majority of cells when production was executed. It cannot be determined when a translocation event in a cell might have carried the transgene structures together with neighboring chromatin from the original chromosomal site to another chromosome (the P chromosome in
Figure 4), generating the indicated PA-1 chromosome. Eventually, possibly favored by a modified culture condition—such as the replacement of FBS-containing media to a serum-free medium—an enrichment of cells carrying the unusual chromosome occurred, likely due to an improved growth rate of cells carrying this unique and new structure. A simplified diagram explaining the possible genetic events (chromosome duplication, translocation), based on the published data, is shown in
Figure 4.
The emergence of a detectable number of cells with the altered chromosomal structure was occurring in a time frame of less than two months of cultures.
All examples provided above indicate massive genome mutations. Unfortunately, so far little information has been established for smaller sized mutations (point mutations, indels, etc.). Such work has apparently only recently been established. Feichinger and co-workers [
23] applying genomic and epigenetic approaches for sequencing with six related CHO cell lines, began to analyze these in more detail and came to the conclusion that the ‘genome homogeneity’ of pools of cells and cloned cell populations do not differ—i.e., cloning seems not to increase overall genetic homogeneity. The analysis is not sufficient in resolution—and probably too complex to execute—to conclude whether any of the observed point mutations have an effect on the physiology of cells in culture.
6. The CHO Quasi-Species Concept
Based on these and other insights the first author of this article proposed several years ago to consider CHO cells as a Quasi-Species family [
24]. The Quasi-Species model, proposed by M. Eigen and P. Schuster [
25] 40 years ago, refers to information carrying systems that undergo self-replication with high error rates. A Quasi-Species is a replicating steady-state population composed of individuals (cells, viruses, nucleic acids) generated with mutations from a Master Sequence. The first sequence for the emergence of a CHO-Quasi-Species is the Hamster genome. The diploid cell that gave rise to subsequent generations of immortalized cells is however not to be considered a true Quasi-Species since that cell had a very low mutation rate (errors) due to the constraints by diploidy, by efficient mutation (allelic) compensation and elaborate DNA repair mechanisms and, in prior replications in the Hamster, by sex-mediated reassembly and functionality requirements. Following the immortalization event, CHO-ori cells (and all subsequently emerging cell lines) were released from diploidy control, and would not need anymore large segments of genomic sequences, required in the animal. Therefore, a Hamster-genome-related but massively modified Quasi-Species evolved that continued to have a high propensity for continual change in DNA and in gene expression profiles.
A key question for our industry therefore is: How can we generate and maintain stable cell lines? Following the concept of Eigen and Schuster, a stable cell line can be maintained only when environmental conditions are kept within narrow and favorable (fitness) ranges that have little impact for the selection of subpopulations of cells (steady state). Under such a condition, the expected genomic sequence variation of CHO cells should be centered around a “stable“ cloud of CHO Master Sequences (CHO-MS) with the highest environmental fitness. Changing media, modifying conditions for subcultivation, periods of diversion from preferred conditions (low oxygen, high or low pH, osmolarity, multiple lag-phases with high waste concentrations, etc.) will contribute to and possibly select for variant genotypes, different from the “averaged” CHO-MS dominating the population of cells before.
7. Cloning and Genetic Stability of Recombinant Cells Expressing Proteins at High Rate
A core concept of ‘cloning’ is that the genetic features of a given clone will be inherited unchanged or nearly unchanged to the progeny of the cloned system, benefiting from advantageous features identified in the clone. In agriculture (wine, apples, etc.) this concept has been the successful basis for food production with identifiable benefit for human consumption for hundreds of years. Besides the necessary care for the cloned plant, no genetic instability is associated with this type of cloning. In E. coli bacteria the technique is applied widely for the multiplication of nucleic acid sequences with the help of extra-chromosomal plasmids. However, due to inherent instability of plasmids in bacteria, the addition of a selection marker and the subsequent use of an antibiotic in culture remains necessary to provide genetic stability in this case.
In animal cells that carry the desired transgene of interest within its chromosomal DNA, the use of a selection marker is not obligatory and frequently not necessary. As the example with methotrexate shows, the presence of this selective agent promotes genetic instability and maintains genetic diversity—different from the clonal cell line identified at the start of the effort. In our view, the counterproductive use—and also the removal of prior use—of selective agents for stability has not been widely recognized by the researchers using CHO or other immortalized cells. The fundamental difference between prokaryotes (extra-chromosomal plasmid DNA) and eukaryotes (genomic intra-chromosomal) in terms of the deposition of transgenic DNA in these organisms has not been taken into consideration. In order to reduce genetic instability trends, clonal or non-clonal cell populations should be ‘allowed’ to recover from any type of selection following transfection and/or the exposure to gene amplifying drugs. In population genetics, the term “population or genetic bottleneck” is used for these periods. Cloning is of course the most serious population bottleneck possible. The recovery periods after such bottlenecks should last as long as 5–10 subcultivations and possibly much more.
Another point to consider is the desire to obtain the highest specific productivity from clonally derived cell populations. However, growth rates of recombinant cells and specific productivity rates are often inversely related to each other. Thus an early evaluation of a clonal population may give an excellent result for its productivity (at small scale). Initially, the emerging population remains closely related to the CHO MS of one member of a prior CHO Quasi-Species family (the clonal cell), possibly chosen for its superior specific productivity, but it may have a non-satisfying growth rate. The time frames necessary to expand this early population for Master Cell Bank generation, seed train cultures, scale-up cultures, and subsequent production phases are very substantial. During these timeframes, a number of different culture conditions are applied and also different media compositions will be used—before the cells arrive in a fully controlled stirred tank bioreactor with pH, oxygen, osmolarity, etc.—all under scrutiny of quality by design principles. During these phases, the members of this new CHO Quasi-Species family (the cells) are evolving and could drift away from the initial CHO MS. cGMP-imposed stability studies, executed from the Master Cell Bank over several months, provides some insights into such trends, but surely will not prevent them.
In our laboratory, we found an example that showed a similar phenomenon. We had established clonally derived cell populations that were grown first in a standard animal component free, suspension culture medium for transfection and clonal selection. Later, we identified for one particular clonal population a medium which seemed to provide overall better productivity. When executing a seed-train based stability study (in the absence of selective conditions) over three months of culture, we observed a trend towards higher cell density and eventually faster growth rates. Better growth, a desired feature of cells, resulted in increasing volumetric productivity over time, while also observing slightly reduced specific productivities over time. Our conclusion from these data, so far unpublished, was that at least one (possibly more) subpopulation of a better fitting CHO Quasi-Species family with a somewhat reduced expression level, but higher growth rate, was over time beginning to dominate the cell population with which the stability study was started. Thus the cell population at the END of the stability study would most likely be composed of a new and better-adapted, “fitter” CHO Quasi-Species population.
It becomes clear then that the first CHO Master Sequence may provide for a population of cells not necessarily most suitable for robust growth or not fully ‘adapted’ to the medium utilized for scale up and manufacturing. The subsequent time frames in culture are more than sufficient to select for alternate expression profiles (and their corresponding mutations genetically) that can improve the growth of the resulting cells. Thus, in spite of eventually having ‘proof’ of clonality the approach does not assure or enhance genetic and phenotypic stability.
8. Conclusions and Recommendations
Identical genetics among the cells of a cell population derived from a single cell (a CHO clone) is not supported by any data. DNA sequence mutations and gross-genomic structure mutations are emerging rapidly (We are discussing mutations that very rarely affect the sequences of the protein of interest, but genomic wide mutations, general point mutations, chromosomal rearrangements, etc.). There are no data to demonstrate that cloning enhances the genetic homogeneity of emergent cell populations. Each clonally-derived population is significantly different from the next one and will present only an averaged genomic sequence composition (‘cloud’) that is expected to be centered around the individual CHO Master Sequence present in the CHO cell deposited into the cloning well. This sequence, as carried forward into multiple replication cycles within the emerging cell population, will evolve further and can either remain closely related to the first CHO MS or evolve into another cloud of sequences that demonstrate better fit to the environments used during the later culture steps. Therefore, culture from a vial of frozen cells towards a production vessel represents an experiment in success-based evolution: Some, hopefully most, emerging cells of the very first thrive in the various environments (or are robust enough to survive them) and will pass their genetic structure (in the form of chromosomes) and DNA content (in terms of sequence composition around the transgene, undisturbed in terms of transcription) to cells that make it into the final production vessel. Actually, many cells with lesser-fit progeny will not end up in the final bioreactor. In any circumstance, and under most efficient processing of cells from a MCB to a 1000 Liter production vessel (typically 30 days) only a small part (estimated as less than 1%) of the theoretical biomass of cells emerging from a vial of frozen cells in a Master Cell Bank will be composed of a cell population structurally highly related and with transgene DNA identical to cells of the original frozen MCB vial.
Cell line stability studies, as a tool to assess the overall performance of cells for manufacturing, have been implemented into research and development efforts of CHO and other cell lines since the first time such cells have been used, more than 30 years ago.
These studies have to be planned and executed very carefully. This is particularly necessary in view of the fact that media, typically animal component free or chemically defined today, can promote much better growth than was the case in the 1980s and 1990s. Also, today a larger offer for such media is available. Stability studies need to be done with clonally derived cell lines that have been maintained ‘sufficiently’ long in a medium intended for manufacturing. It is preferred to have a medium that ‘matches’ to the averaged physiology, based on the transcription from the cloud of sequences around the CHO MS (the “fittest” in terms of match to the medium). The apparent preference of the industry to apply one medium or a few platform media for many cell lines is ill-advised. Of course, considering the enormous genetic and physiological responsiveness of CHO populations, eventually and probably relatively fast, cells will “adapt” almost to any of the chosen media, but the consequences of bottlenecking populations are unpredictable, as discussed and shown in the examples provided in this paper.
It is difficult to state here how many sub-cultivations, and thus number of population doublings, using a chosen manufacturing medium are necessary to achieve a level of steady-state in the emerged cell population. We believe however it should be at least 10 sub-cultivations, preferably under conditions that closely match the sub-cultivation steps (including the ratios of sub-cultivations and seed densities for the subsequent culture phase) executed eventually during scale up towards manufacturing.
The question of selective agents in media is more complicated. In view of the observations discussed above, preferably drug-free seed train cultures and scale-up cultures should be used for manufacturing. However, there may be products and their corresponding manufacturing processes that have been established with cell cultures maintained under selection pressure. The authors recommend in such a case additional studies that show the performance of cells in the absence of selection drugs, in order to begin to understand the complex population genetics of such cells.
Yet another important consideration, but still in connection with the chosen medium, is the general physico-chemical environment in which cells are propagated. Culture steps towards the largest bioreactor—the final production vessel, will differ more or less, depending on the technology used, from the environment in which seed train cultures are executed. Typically, for scale up, when done in fully controlled bioreactors with a high degree of control over key parameters—such as oxygen supply and/or pH range—the environment will be a “less selective” one than those used in stability studies (Static or shaken multi-well plates or Erlenmeyer type flasks). In these systems, gas transfer rates and mixing are low and/or poorly studied. The more variable conditions concerning pH, oxygen-supply, dissolved CO2 concentration can affect the cultures in non-predictable ways.
Even for “controlled” reactors, some new systems such as the widely used “Wave type” present a dilemma. These vessels have entirely different fluid-dynamics from stirred tanks, and their oxygen transfer is rather low, necessitating the use of pure oxygen. Unfortunately, we know of no experimental work that has been performed on how different chemical engineering parameters affect the biology of heterogenetic cell populations.
Overall, while the public perceives mammalian cell culture-based manufacturing for biopharmaceuticals an ‘established’ technology, the authors of this paper—having worked in this field cumulatively now for more than 60 years—feel humbled by the complexities observed and the many open questions remaining to be solved for the future.



{kind=link}
{kind=link}
{kind=link}
{kind=link}



