Systematic Selection of Green Solvents and Process Optimization for the Hydroformylation of Long-Chain Olefines

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Solvent Screening
2.2. Process Model
2.2.1. Reactor
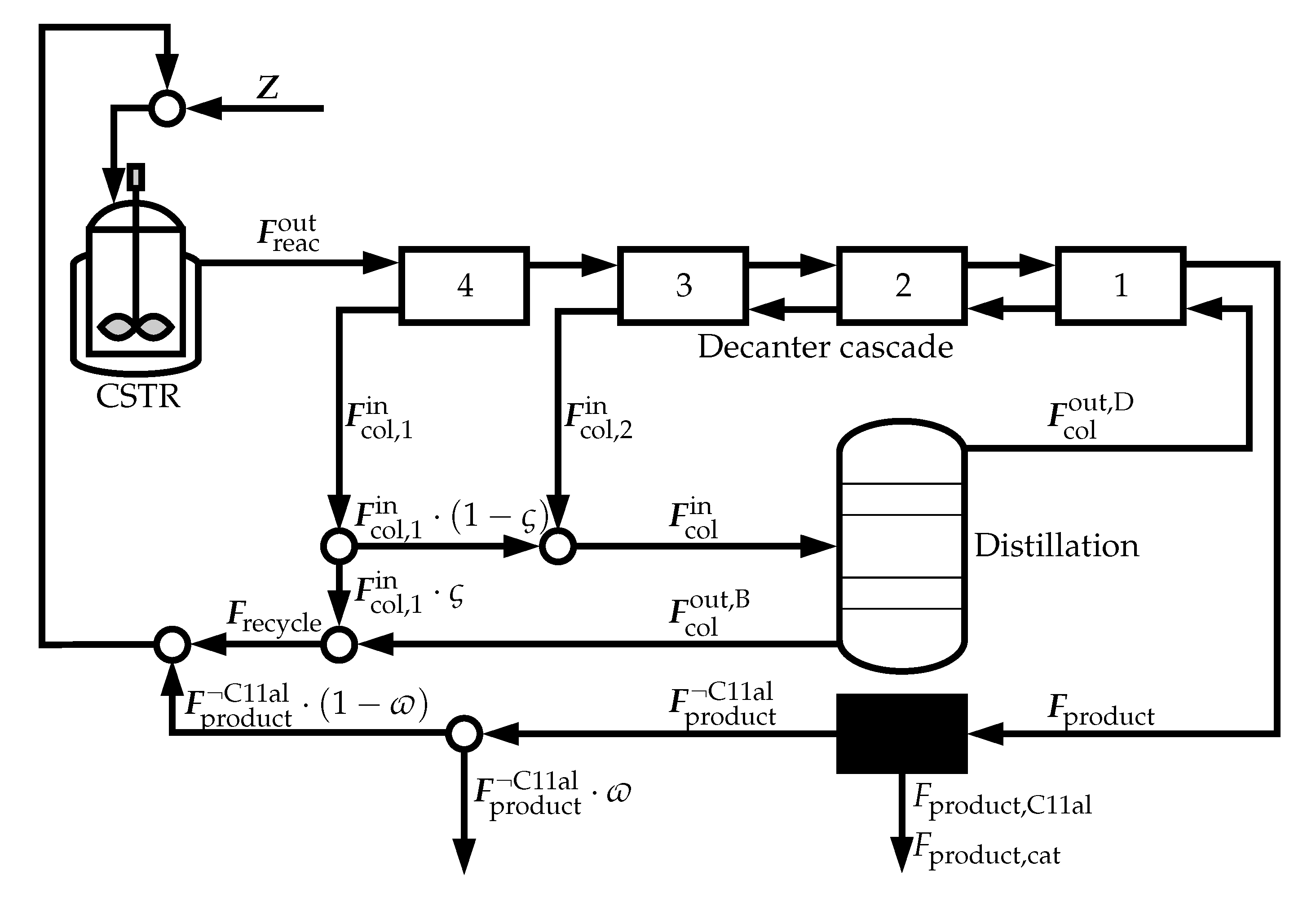
2.2.2. Downstream Processing
- DMF: 99% purity of DMF in the distillate
- DSUC: 99% purity of DSUC + C10en in the distillate
- THPO: 99% purity of THPO in the distillate
2.2.3. Black Box
2.2.4. Overall Process
2.3. Process Optimization
3. Results
3.1. Process Optimization for Each Candidate Solvent
3.1.1. Dimethylformamide (DMF)
3.1.2. Dimethyl Succinate (DSUC)
3.1.3. Tetrahydropyranone (THPO)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Nomenclature
| Vector-valued: | |
| vapor pressure parameters | |
| relative volatilities | |
| mean temperature | |
| liquid mole fraction | |
| vector of activity coefficients | |
| cost function parameters | |
| column cost parameters | |
| stoichiometric coefficients | |
| vapor mole fraction | |
| concentration | |
| molar flow rate | |
| inequality constraints | |
| equality constraints | |
| reaction rate parameters | |
| reaction rate constants | |
| molar flow rate, phase I | |
| vapor pressure | |
| reaction rates | |
| molar flow rate, phase II | |
| liquid composition, phase I | |
| liquid composition, phase II | |
| molar feed flow rate | |
| Real-valued: | |
| purge fraction | |
| split fraction | |
| count variable | |
| objective function | |
| column length | |
| P | pressure |
| polar solvent | |
| T | temperature |
| total annualized cost | |
| vapor flow rate, column | |
| reactor volume | |
| Abbreviations: | |
| ANN | Artificial Neural Network |
| CAMD | Computer-Aided Molecular Design |
| COSMO-RS | Continuum Solvation Model for Realistic Solvents |
| DMF | Dimethylformamide |
| DSUC | Dimethylsuccinate |
| EHS | Environment, health, and safety |
| EOS | Equation Of State |
| FUG | Fenske–Underwood–Gilliland |
| InPROMPT | Integrated chemical processes in liquid multiphase system |
| LLE | Liquid–Liquid Equilibrium |
| MINLP | Mixed-Integer Non-Linear Program |
| MOO | Multi-Objective Optimization |
| PC-SAFT | Perturbed-Chain Statistical Associating Fluid Theory |
| RA | Reductive Amination |
| SVHC | Substance of Very High Concern |
| THPO | Tetrahydropyranone |
| TMS | Thermomorphic Multiphase System |
| UNIFAC | Universal Quasichemical Functional Group Activity Coefficients |
Appendix A

{kind=link}
{kind=link}
| a | a | a | a | a | |
|---|---|---|---|---|---|
| DMF | −47.9857 | × | 28.8 | × | × |
| DSUC | 117.8014 | × | −42.5731 | × | × |
| THPO | 74.2227 | × | −25.9627 | × | × |
| C10en | 2.2678 | × | 5.43 | × | × |
| C12an | −8.5899 | × | × | × | |
| C11al | −31.8129 | × | 20.4 | × | × |
| Parameter | DMF | DSUC | THPO |
|---|---|---|---|
| 15,071 | 17,783 | ||
| 2988.6 | |||
| 1 | 1 | 1 | |
| 202.9541 | |||
| 1 | 1 | 1 | |
| 64,191 | 168,833 | 51,628 | |
| 20,638 | 19,656 | 20,695 | |
| −56,984 | −182,134 | −53,368 | |
| −9.6219 | −4.2671 | −12.5046 | |
| 98,841 | 439,840 | 71,678 | |
| × | × | × | |
| × | × | × | |
| × | × | × | |
| × | × | × | |
| 504,155 | 504,155 | 504,155 | |
| 0.586667 | 0.586667 | 0.586667 | |
| × | × | × | |
| 85,150 | 85,150 | 85,150 | |
| 17,248.42 | 17,248.42 | 17,248.42 | |
| 0.62 | 0.62 | 0.62 | |
| × | × | × | |
| 0.661 | 0.661 | 0.661 | |
| 0.0731 | 0.0731 | 0.0731 | |
| 0.0714 | 0.0714 | 0.0714 |
| DMF | 31.9942 | [-] | [-] |
| DMS | [-] | 4.0208 | [-] |
| THPO | [-] | [-] | 22.4438 |
| C12an | 3.1307 | 2.7766 | 3.0696 |
| C10en | 16.8669 | 11.7308 | 15.8739 |
| C11al | 1 | 1 | 1 |
References
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Linthorst, J.A. An overview: Origins and development of green chemistry. Found. Chem. 2010, 12, 55–68. [Google Scholar] [CrossRef]
- Hoffenson, S.; Dagman, A.; Söderberg, R. Re-engineering Manufacturing for Sustainability; Chapter A Multi-Objective Tolerance Optimization Approach for Economic, Ecological, and Social Sustainability; Springer: Berlin, Germany, 2013; pp. 729–734. [Google Scholar]
- Elkington, J. Cannibals with Forks: The Triple Bottom Line of 21st Century Business; New Society Publishers: Gabriola Island, BC, Canada, 1998. [Google Scholar]
- Lundberg, I.; Lidén, C. Handbook of Hazardous Materials; chapter Industrial Solvents; Academic Press: San Diego, CA, USA, 1993; pp. 387–397. [Google Scholar] [CrossRef]
- Joback, K.G.; Reid, R.C. Estimation of Pure-Component Properties from Group-Contributions. Chem. Eng. Commun. 1987, 56, 233–243. [Google Scholar] [CrossRef]
- Burger, J.; Papaioannou, V.; Gopinath, S.; Jackson, G.; Galindo, A.; Adjiman, C.S. A hierarchical method to integrated solvent and process design of physical CO2 absorption using the SAFT-γ Mie approach. AIChE J. 2015, 61, 3249–3269. [Google Scholar] [CrossRef]
- Papadopoulos, A.I.; Stijepovic, M.; Linke, P. On the systematic design and selection of optimal working fluids for Organic Rankine Cycles. Appl. Therm. Eng. 2010, 30, 760–769. [Google Scholar] [CrossRef]
- Zhou, T.; Song, Z.; Zhang, X.; Gani, R.; Sundmacher, K. Optimal Solvent Design for Extractive Distillation Processes: A Multiobjective Optimization-Based Hierarchical Framework. Ind. Eng. Chem. Res. 2019, 58, 5777–5786. [Google Scholar] [CrossRef]
- Karunanithi, A.T.; Achenie, L.E.K.; Gani, R. A New Decomposition-Based Computer-Aided Molecular/Mixture Design Methodology for the Design of Optimal Solvents and Solvent Mixtures. Ind. Eng. Chem. Res. 2005, 44, 4785–4797. [Google Scholar] [CrossRef]
- Cignitti, S.; Mansouri, S.S.; Woodley, J.M.; Abildskov, J. Systematic Optimization-Based Integrated Chemical Product–Process Design Framework. Ind. Eng. Chem. Res. 2017, 57, 677–688. [Google Scholar] [CrossRef]
- Eden, M.; Jøorgensen, S.; Gani, R.; El-Halwagi, M. A novel framework for simultaneous separation process and product design. Chem. Eng. Process. 2004, 43, 595–608. [Google Scholar] [CrossRef]
- Kossack, S.; Kraemer, K.; Gani, R.; Marquardt, W. A systematic synthesis framework for extractive distillation processes. Chem. Eng. Res. Des. 2008, 86, 781–792. [Google Scholar] [CrossRef]
- Gopinath, S.; Jackson, G.; Galindo, A.; Adjiman, C.S. Outer approximation algorithm with physical domain reduction for computer-aided molecular and separation process design. AIChE J. 2016, 62, 3484–3504. [Google Scholar] [CrossRef]
- First, E.L.; Hasan, M.M.F.; Floudas, C.A. Discovery of novel zeolites for natural gas purification through combined material screening and process optimization. AIChE J. 2014, 60, 1767–1785. [Google Scholar] [CrossRef]
- Jens, C.M.; Nowakowski, K.; Scheffczyk, J.; Leonhard, K.; Bardow, A. CO from CO 2 and fluctuating renewable energy via formic-acid derivatives. Green Chem. 2016, 18, 5621–5629. [Google Scholar] [CrossRef]
- Zhou, T.; Zhou, Y.; Sundmacher, K. A hybrid stochastic–deterministic optimization approach for integrated solvent and process design. Chem. Eng. Sci. 2017, 159, 207–216. [Google Scholar] [CrossRef]
- Papadopoulos, A.I.; Linke, P. Multiobjective molecular design for integrated process-solvent systems synthesis. AIChE J. 2005, 52, 1057–1070. [Google Scholar] [CrossRef]
- Limleamthong, P.; Gonzalez-Miquel, M.; Papadokonstantakis, S.; Papadopoulos, A.I.; Seferlis, P.; Guillén-Gosálbez, G. Multi-criteria screening of chemicals considering thermodynamic and life cycle assessment metrics via data envelopment analysis: Application to CO2 capture. Green Chemistry 2016, 18, 6468–6481. [Google Scholar] [CrossRef]
- Ten, J.Y.; Hassim, M.H.; Ng, D.K.S.; Chemmangattuvalappil, N.G. A molecular design methodology by the simultaneous optimisation of performance, safety and health aspects. Chem. Eng. Sci. 2017, 159, 140–153. [Google Scholar] [CrossRef]
- Scheffczyk, J.; Redepenning, C.; Jens, C.M.; Winter, B.; Leonhard, K.; Marquardt, W.; Bardow, A. Massive, automated solvent screening for minimum energy demand in hybrid extraction–distillation using COSMO-RS. Chem. Eng. Res. Des. 2016, 115, 433–442. [Google Scholar] [CrossRef]
- Scheffczyk, J.; Schäfer, P.; Fleitmann, L.; Thien, J.; Redepenning, C.; Leonhard, K.; Marquardt, W.; Bardow, A. COSMO-CAMPD: A framework for integrated design of molecules and processes based on COSMO-RS. Mol. Syst. Des. Eng. 2018, 3, 645–657. [Google Scholar] [CrossRef]
- Scheffczyk, J.; Schäfer, P.; Jens, C.M.; Leonhard, K.; Bardow, A. Integrated process and solvent design using COSMO-RS for the production of CO from CO2 and H2. In Computer Aided Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2017; Volume 40, pp. 1765–1770. [Google Scholar] [CrossRef]
- Fleitmann, L.; Scheffczyk, J.; Schäfer, P.; Jens, C.M.; Leonhard, K.; Bardow, A. Integrated Design of Solvents in Hybrid Reaction-Separation Processes Using COSMO-RS. Chem. Eng. Trans. 2018, 2018, 559–564. [Google Scholar] [CrossRef]
- Bardow, A.; Steur, K.; Gross, J. Continuous-Molecular Targeting for Integrated Solvent and Process Design. Ind. Eng. Chem. Res. 2010, 49, 2834–2840. [Google Scholar] [CrossRef]
- Stavrou, M.; Lampe, M.; Bardow, A.; Gross, J. Continuous Molecular Targeting–Computer-Aided Molecular Design (CoMT–CAMD) for Simultaneous Process and Solvent Design for CO2 Capture. Ind. Eng. Chem. Res. 2014, 53, 18029–18041. [Google Scholar] [CrossRef]
- Wang, J.; Lakerveld, R. Integrated solvent and process design for continuous crystallization and solvent recycling using PC-SAFT. AIChE J. 2018, 64, 1205–1216. [Google Scholar] [CrossRef]
- Lampe, M.; Stavrou, M.; Schilling, J.; Sauer, E.; Gross, J.; Bardow, A. Computer-aided molecular design in the continuous-molecular targeting framework using group-contribution PC-SAFT. Comput. Chem. Eng. 2015, 81, 278–287. [Google Scholar] [CrossRef]
- McBride, K.; Linke, S.; Xu, S.; Sundmacher, K. Computer Aided Design of Green Thermomorphic Solvent Systems for Homogeneous Catalyst Recovery. In Proceedings of the 13th International Symposium on Process Systems Engineering (PSE 2018), San Diego, CA, USA, 1–5 July 2018; pp. 1783–1788. [Google Scholar] [CrossRef]
- Bianga, J.; Künnemann, K.; Gaide, T.; Vorholt, A.J.; Seidensticker, T.; Dreimann, J.; Vogt, D. Thermomorphic Multiphase Systems: Switchable Solvent Mixtures for the Recovery of Homogeneous Catalysts in Batch and Flow Processes. Chem. Eur. J. 2019, 25, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, E.; Brunsch, Y.; Sadowski, G.; Behr, A. Hydroformylation of 1-Dodecene in the Thermomorphic Solvent System Dimethylformamide/Decane. Phase Behavior-Reaction Performance-Catalyst Recycling. Ind. Eng. Chem. Res. 2012, 51, 10296–10306. [Google Scholar] [CrossRef]
- Kiedorf, G.; Hoang, D.; Müller, A.; Jörke, A.; Markert, J.; Arellano-Garcia, H.; Seidel-Morgenstern, A.; Hamel, C. Kinetics of 1-dodecene hydroformylation in a thermomorphic solvent system using a rhodium-biphephos catalyst. Chem. Eng. Sci. 2014, 115, 31–48. [Google Scholar] [CrossRef]
- Hentschel, B.; Kiedorf, G.; Gerlach, M.; Hamel, C.; Seidel-Morgenstern, A.; Freund, H.; Sundmacher, K. Model-Based Identification and Experimental Validation of the Optimal Reaction Route for the Hydroformylation of 1-Dodecene. Ind. Eng. Chem. Res. 2015, 54, 1755–1765. [Google Scholar] [CrossRef]
- Keßler, T.; Mertens, N.; Kunde, C.; Nentwich, C.; Michaels, D.; Engell, S.; Kienle, A. Efficient global optimization of a novel hydroformylation process. In 27th European Symposium on Computer Aided Process Engineering; Espuña, A., Graells, M., Puigjaner, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 40, pp. 2113–2118. [Google Scholar] [CrossRef]
- McBride, K.; Sundmacher, K. Data Driven Conceptual Process Design for the Hydroformylation of 1-Dodecene in a Thermomorphic Solvent System. Ind. Eng. Chem. Res. 2015, 54, 6761–6771. [Google Scholar] [CrossRef]
- Nentwich, C.; Engell, S. Application of surrogate models for the optimization and design of chemical processes. In Proceedings of the 2016 International Joint Conference on Neural Networks (IJCNN), Vancouver, BC, Canada, 24–29 July 2016; pp. 1291–1296. [Google Scholar] [CrossRef]
- Dreimann, J.M.; Hoffmann, F.; Skiborowski, M.; Behr, A.; Vorholt, A.J. Merging Thermomorphic Solvent Systems and Organic Solvent Nanofiltration for Hybrid Catalyst Recovery in a Hydroformylation Process. Ind. Eng. Chem. Res. 2017, 56. [Google Scholar] [CrossRef]
- Hernández, R.; Engell, S. Modelling and iterative real-time optimization of a homogeneously catalyzed hydroformylation process. Comput. Aided Chem. Eng. 2016, 38, 1–6. [Google Scholar] [CrossRef]
- Illner, M.; Müller, D.; Esche, E.; Pogrzeba, T.; Schmidt, M.; Schomäcker, R.; Wozny, G.; Repke, J.U. Hydroformylation in Microemulsions: Proof of Concept in a Miniplant. Ind. Eng. Chem. Res. 2016, 55. [Google Scholar] [CrossRef]
- Müller, D.; Illner, M.; Esche, E.; Pogrzeba, T.; Schmidt, M.; Schomäcker, R.; Biegler, L.T.; Wozny, G.; Repke, J.U. Dynamic real-time optimization under uncertainty of a hydroformylation mini-plant. Comput. Chem. Eng. 2017, 106, 836–848. [Google Scholar] [CrossRef]
- Kraume, M. Integrierte chemische Prozesse in flüssigen Mehrphasensystemen. Chemie Ingenieur Technik 2013, 85, 1499–1511. [Google Scholar] [CrossRef]
- Behr, A.; Vorholt, A.J. Hydroformylation and Related Reactions of Renewable Resources; Springer: Berlin/Heidelberg, Germany, 2012; pp. 103–127. [Google Scholar] [CrossRef]
- Behr, A.; Obst, D.; Westfechtel, A. Isomerizing hydroformylation of fatty acid esters: Formation of ω-aldehydes. Eur. J. Lipid Sci. Technol. 2005, 107, 213–219. [Google Scholar] [CrossRef]
- Fail, P.A.; George, J.D.; Grizzle, T.B.; Heindel, J.J. Formamide and Dimethylformamide: Reproductive Assessment by Continuous Breeding in Mice. Reprod. Toxicol. 1998, 12. [Google Scholar] [CrossRef]
- Kleiner, D.E. Macsween’s Pathology of the Liver; Chapter 12—Drugs and Toxins; Elsevier: Amsterdam, The Netherlands, 2018; pp. 673–779. [Google Scholar] [CrossRef]
- Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH). Available online: https://echa.europa.eu (accessed on 4 September 2019).
- Klamt, A.; Jonas, V.; Bürger, T.; Lohrenz, J.C.W. Refinement and Parametrization of COSMO-RS. J. Phys. Chem. A 1998, 102, 5074–5085. [Google Scholar] [CrossRef]
- COSMOtherm, C30; Release 1601; COSMOlogic GmbH & Co. KG: Leverkusen, Germany, 2016; Available online: http://www.cosmologic.de (accessed on 25 November 2019).
- McBride, K.; Gaide, T.; Vorholt, A.; Behr, A.; Sundmacher, K. Thermomorphic solvent selection for homogeneous catalyst recovery based on COSMO-RS. Chem. Eng. Process. 2016, 99, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Benfenati, E.; Manganaro, A.; Gini, G. VEGA-QSAR: AI inside a platform for predictive toxicology. In CEUR Workshop Proceedings; CEUR-WS: Turin, Italy, 5 December 2013; Volume 1107, pp. 21–28. [Google Scholar]
- US EPA. Estimation Programs Interface Suite for Microsoft Windows (EPISuite). 2017. Available online: https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface (accessed on 25 November 2019).
- Moity, L.; Durand, M.; Benazzouz, A.; Pierlot, C.; Molinier, V.; Aubry, J.M. Panorama of sustainable solvents using the COSMO-RS approach. Green Chem. 2012, 14, 1132. [Google Scholar] [CrossRef]
- Kılınç, M.R.; Sahinidis, N.V. Exploiting integrality in the global optimization of mixed-integer nonlinear programming problems with BARON. Optim. Methods Softw. 2018, 33, 540–562. [Google Scholar] [CrossRef]
- Henley, E.J.; Seader, J.D. Equilibrium-Stage Separation Operations in Chemical Engineering; John Wiley & Sons, Inc.: New York, NY, USA, 1981. [Google Scholar]
- Keßler, T.; Kunde, C.; McBride, K.; Mertens, N.; Michaels, D.; Sundmacher, K.; Kienle, A. Global optimization of distillation columns using explicit and implicit surrogate models. Chem. Eng. Sci. 2019, 197, 235–245. [Google Scholar] [CrossRef]
- McBride, K.; Kaiser, N.M.; Sundmacher, K. Integrated reaction-extraction process for the hydroformylation of long-chain alkenes with a homogeneous catalyst. Comput. Chem. Eng. 2017, 105, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Dreimann, J.M.; Warmeling, H.; Weimann, J.N.; Künnemann, K.; Behr, A.; Vorholt, A.J. Increasing selectivity of the hydroformylation in a miniplant: Catalyst, solvent, and olefin recycle in two loops. AIChE J. 2016, 62, 4377–4383. [Google Scholar] [CrossRef]
- Weidlich, U.; Gmehling, J. A modified UNIFAC model. 1. Prediction of VLE, hE and γ∞. Ind. Eng. Chem. Res. 1987, 26, 1372–1381. [Google Scholar] [CrossRef]
- Chen, C.C.; Song, Y. Solubility Modeling with a Nonrandom Two-Liquid Segment Activity Coefficient Model. Ind. Eng. Chem. Res. 2004, 43, 8354–8362. [Google Scholar] [CrossRef]
- Seydel, R. Practical Bifurcation and Stability Analysis; Springer: Berlin, Germany, 2009. [Google Scholar] [CrossRef]
- Kunde, C.; Keßler, T.; Linke, S.; McBride, K.; Sundmacher, K.; Kienle, A. Surrogate modeling for liquid–liquid equilibria using a parametrization of the binodal curve. Processes 2019, 7, 753. [Google Scholar] [CrossRef] [Green Version]
- Moller, B.; Rarey, J.; Ramjugernath, D. Estimation of the vapour pressure of non-electrolyte organic compounds via group contributions and group interactions. J. Mol. Liq. 2008, 143, 52–63. [Google Scholar] [CrossRef]
- Yaws, C. Chemical Properties Handbook: Physical, Thermodynamics, Engironmental Transport, Safety & Health Related Properties for Organic & Inorganic Chemical; McGraw-Hill Education: Berkshire, UK, 1998. [Google Scholar]


| Index | Name | Purpose | Produced by |
|---|---|---|---|
| C10en | n-decene | reactant | |
| iC10en | iso-decene | side product | isomerisation of n-decene (reversible) |
| C11al | n-undecanal | desired product | hydroformylation of n-decene (irreversible) |
| iC11al | iso-undecanal | side product | hydroformylation of iso-decene (irreversible) |
| C12an | n-dodecane | non-polar solvent | |
| DMF | dimethylformamide | polar solvent | |
| DSUC | dimethyl succinate | polar solvent | |
| THPO | tetrahydropyranone | polar solvent | |
| C10an | n-decane | side product | hydrogenation of n-/iso-decene (irreversible) |
| RHO | rhodium | catalyst | |
| BIP | biphephos | ligand | |
| H | hydrogen | reactant | |
| CO | carbon monoxide | reactant |
| Polar Solvent | Volume (m) | Reactor Pressure (bar) | Temperature (K) | Recycle Purge (-) | Extraction Cascade Split Factor (-) | Overall Process Costs ($/a) |
|---|---|---|---|---|---|---|
| DMF | 273.3775 | 20 | 388.15 | 0.0013 | 0 | 1,934,522 (100 %) |
| DSUC | 3458.1775 | 20 | 388.15 | 0.0018 | 0 | 6,154,228 (+218 %) |
| THPO | 203.8447 | 20 | 388.15 | 0.0015 | 0 | 1,748,021 (−9.64 %) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keßler, T.; Kunde, C.; Linke, S.; McBride, K.; Sundmacher, K.; Kienle, A. Systematic Selection of Green Solvents and Process Optimization for the Hydroformylation of Long-Chain Olefines. Processes 2019, 7, 882. https://doi.org/10.3390/pr7120882
Keßler T, Kunde C, Linke S, McBride K, Sundmacher K, Kienle A. Systematic Selection of Green Solvents and Process Optimization for the Hydroformylation of Long-Chain Olefines. Processes. 2019; 7(12):882. https://doi.org/10.3390/pr7120882
Chicago/Turabian StyleKeßler, Tobias, Christian Kunde, Steffen Linke, Kevin McBride, Kai Sundmacher, and Achim Kienle. 2019. "Systematic Selection of Green Solvents and Process Optimization for the Hydroformylation of Long-Chain Olefines" Processes 7, no. 12: 882. https://doi.org/10.3390/pr7120882
APA StyleKeßler, T., Kunde, C., Linke, S., McBride, K., Sundmacher, K., & Kienle, A. (2019). Systematic Selection of Green Solvents and Process Optimization for the Hydroformylation of Long-Chain Olefines. Processes, 7(12), 882. https://doi.org/10.3390/pr7120882