1. Introduction
Polyethylene (PE), one of the most used and commercialized thermoplastics in the world, is produced by the polymerization of ethylene which is catalyzed via two main different routes: using heterogeneous processes with Ziegler–Natta catalysts, or via metallocene catalytic systems. Since the discovery of the catalytic activity of the homogenous catalysts based on biscyclopentadienyl titanium or zirconium dialkyl systems in the ethylene polymerization in the 1980s by Kaminsky and Sinn [
1,
2,
3,
4], metallocene systems have revolutionized the polyolefins field, because they enable the production of PE with narrow molecular weight distributions, low content of extractables, good processability, and superior properties [
5]. Moreover, metallocene catalysts, in comparison to Ziegler–Natta types, show a single type of active site, which enables predictions of the properties of the resulting polymers.
Several factors play an important role in the olefin’s polymerization via metallocene catalysis. For instance, the formation of weakly coordinating anions with a weak bonding to the metallocene active centers (acting as co-catalysts). The anions interact with the cationic metal species, in the reaction medium, creating active sites (ion-pairs), followed by the subsequent polymerization. Methylaluminoxane (MAO) is a popular activator due to its high efficiency; however, a large excess of MAO is usually required, and, despite extensive efforts, its detailed active-site structure has not yet been fully elucidated [
6,
7,
8]. A prominent alternative to replace MAO is the use of other bulky coordinating anions such as organoboranes, e.g., tris(pentafluorophenyl)borane (B1) [
9,
10], and organoborates such as
N,N-dimethylanilinium tetra(pentafluorophenyl)borate (B2) or trityl tetra(pentafluorophenyl)borate (B3) [
11,
12]. These types of activators can ionize the metallocene (pre-alkylated) catalyst, acting as Lewis acids, leading to excellent active cationic metallocene catalysts for the polymerization of olefins in quasi-equimolar amounts between the metallocene catalyst and the boron-based activator, and resulting in catalytic complexes with a definite chemical structure [
13,
14,
15]. A breakthrough in this field was the introduction of the weakly coordinating tris(pentafluorophenyl)borate [B(C
6F
5)
3] as a counterion, which can abstract a methyl group from the alkylated metallocene catalyst, to form ionic species such as [CP
2ZrMe]
+[MeB(C
6F
5)
3]
−, followed by the coordination of a monomer molecule and subsequent propagation [
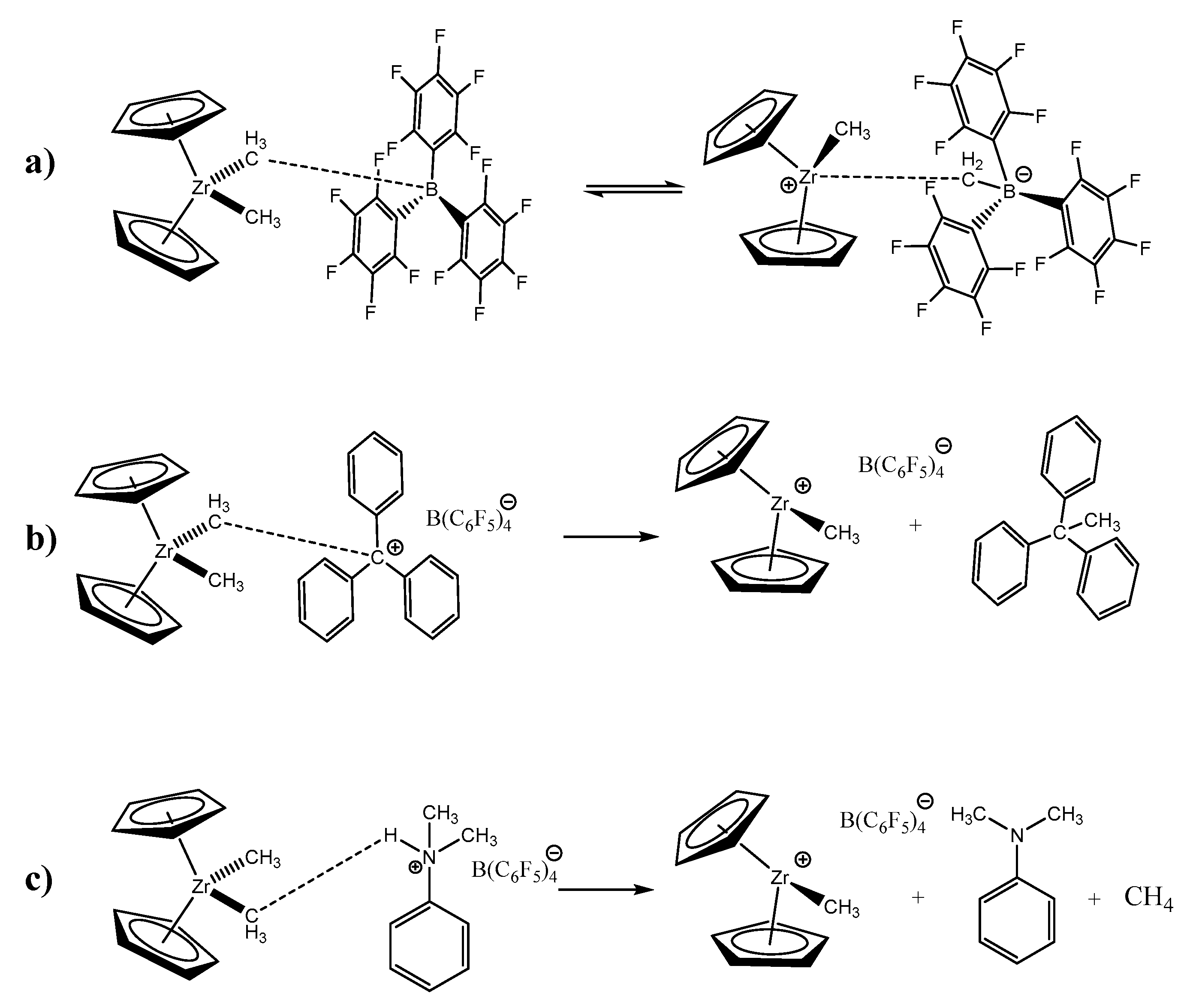
10]. Nevertheless, residual coordinative interactions between the activated metal center and the anion, via the abstracted methyl group, can slightly decrease the catalyst reactivity. Ionic organoboron activators, such as [C
6H
5NHMe
2]
+[B(C
6F
5)
4]
− and [(C
6H
5)
3C]
+[B(C
6F
5)
4]
−, on the other hand, avoid this form of ion–ion contact between the abstracted methyl group and the transition metal atom, while the cationic species takes the function of activating the metal active center (
Figure 1).
There are a wide variety of metallocene catalysts with different symmetries and substitutions, because the configuration of the catalyst is another factor governing polymerization behavior. For instance, the steric and electronic environment of ligand substituents of the metal catalyst, as well as the ion–ion interactions between the electrophilic metal and the counterion, are critical factors that can dramatically alter the polymerization behavior due to steric hindrance and electronic factors.
Several works have previously studied this behavior. For instance, Ewen and Chien [
13,
16], studied the effect of different alkyl substituents in cyclopentadienyl (CP) groups for several zirconocene (Zr) catalysts, reporting the following behavior in terms of catalyst efficiency: (MeCP)
2ZrCl
2 > (EtCP)
2ZrCl
2 > CP
2ZrCl
2 > (Me
5CP)CPZrCl
2. Through this, they concluded that single alkyl substituents increase the catalytic activity due to electro donating effects, while the steric hindrance of bulky substituents has a detrimental effect instead. Zr-based catalysts have been also studied in heterogeneous systems for ethylene polymerizations; for example, Charles et al. reported ethylene polymerization using catalysts derived from the activation of Zr aluminohydride complexes, supported on silica, which was previously treated with MAO. The results were compared with those using the more traditional Zr dichloro complexes, finding higher activity in the former [
17]. Zeolites (ZSM-5) [
18], and solid polymethylaluminoxane [
19] are among the supports reported for carrying out ethylene polymerizations catalyzed by Zr-based metallocenes, achieving high catalytic activities, high molecular weights, and narrow distributions. Although these works provide general features about the influence of the alkyl groups on the ligand substituents, and the influence of using solid supports during the polymerization, they were all carried out using MAO as the activator.
Few works have studied in-detail the ethylene polymerization behavior when the metallocene is activated by the bulky, weakly-coordinating organoboron anions (B). In this sense, our research group reported the use of tris(pentafluorophenyl)borane and
N,N-dimethylanilinium tetrakis(pentafluorophenyl)borate (B1 and B2 in this work, respectively) to act in conjunction with MAO as activators on ethylene polymerization by using the catalyst CP
2ZrCl
2. The addition of these organoboron compounds of ionic and nonionic nature in a molar ratio B1(or B2)/Zr = 5 promoted a partial deactivation of the catalyst, causing a reduction in the catalytic activity; however, the crystallinity degree, as well as the macromolecular, thermal, and dynamic-mechanical properties of the obtained polyethylenes were improved, especially with B1 as co-activator in this evaluated catalytic system [
14]. In the same context, González-Hernández et al. [
19] reported the ethylene polymerization using catalysts derived from Zr aluminohydride complexes activated with tris(pentafluorophenyl)borane (B1), although with limited utility (catalytic activity) of these catalysts systems when compared with the corresponding use of MAO as the activator. Supported zirconocene catalysts activated by boron compounds for olefin polymerizations are not as widely reported in the literature, but there are some related works such as that reported by Charoenchidet et al. who treated silica with tris(pentafluorophenyl)borane (B1 in this work) to produce borane-functionalized support, which was then used as a support and co-catalyst for the CP
2ZrCl
2, CP
2ZrCl
2/Triisobutylaluminum (TIBA), CP
2Zr(CH
3)
2 and CP
2Zr(CH
3)
2/TIBA catalyst systems for ethylene polymerizations. The activations of the catalysts were carried out in two ways: pre-activation, and in situ activation. The pre-activated and in situ-activated metallocene systems produced PE with M
w between 96 and 154 Kg mol
−1, and dispersity index (
Ð) around 3. The bulk density of PE products was higher for the in situ-activated systems, but there was no significant difference between the products of both types of zirconocenes [
20].
On the other hand, the kinetics of the catalyst coordination polymerization has been previously simulated, however a low number of reports can be found, compared to free-radical polymerization systems. Chien and Wang [
13] reported the first kinetic model to study polymerization using zirconocene dichloride (CP
2ZrCl
2) and MAO as the catalyst and co-catalyst, respectively. The kinetic mechanism proposed the chain transfer to MAO, β-hydride chain transfer, multiple types of active sites, and deactivation step. Estrada and Hamielec [
21] developed a model with two types of active sites, where the first one experienced a gradual transition (a state change) to the second type; this step was supported on the bimodal molecular weight distribution observed in the size exclusion chromatography (SEC) measurements. Both models did not provide an estimation of the ethylene concentration in the liquid phase. Moreover, Jiang et al. [
22] carried out a comparative study between different models: in one of them, the reactivation of MAO was included as part of the kinetic mechanism, resulting in better agreement with the experimental polymerization rate profiles. A strategy of parameter estimation was reported by Ahmadi et al., in which a multivariable nonlinear optimization problem was solved using the Nelder–Mead simplex method [
23]. The methodology combined the numerical solution of the kinetic model with the optimization algorithm, resulting in good agreement with the experimental data. Mehdiabadi and Soares [
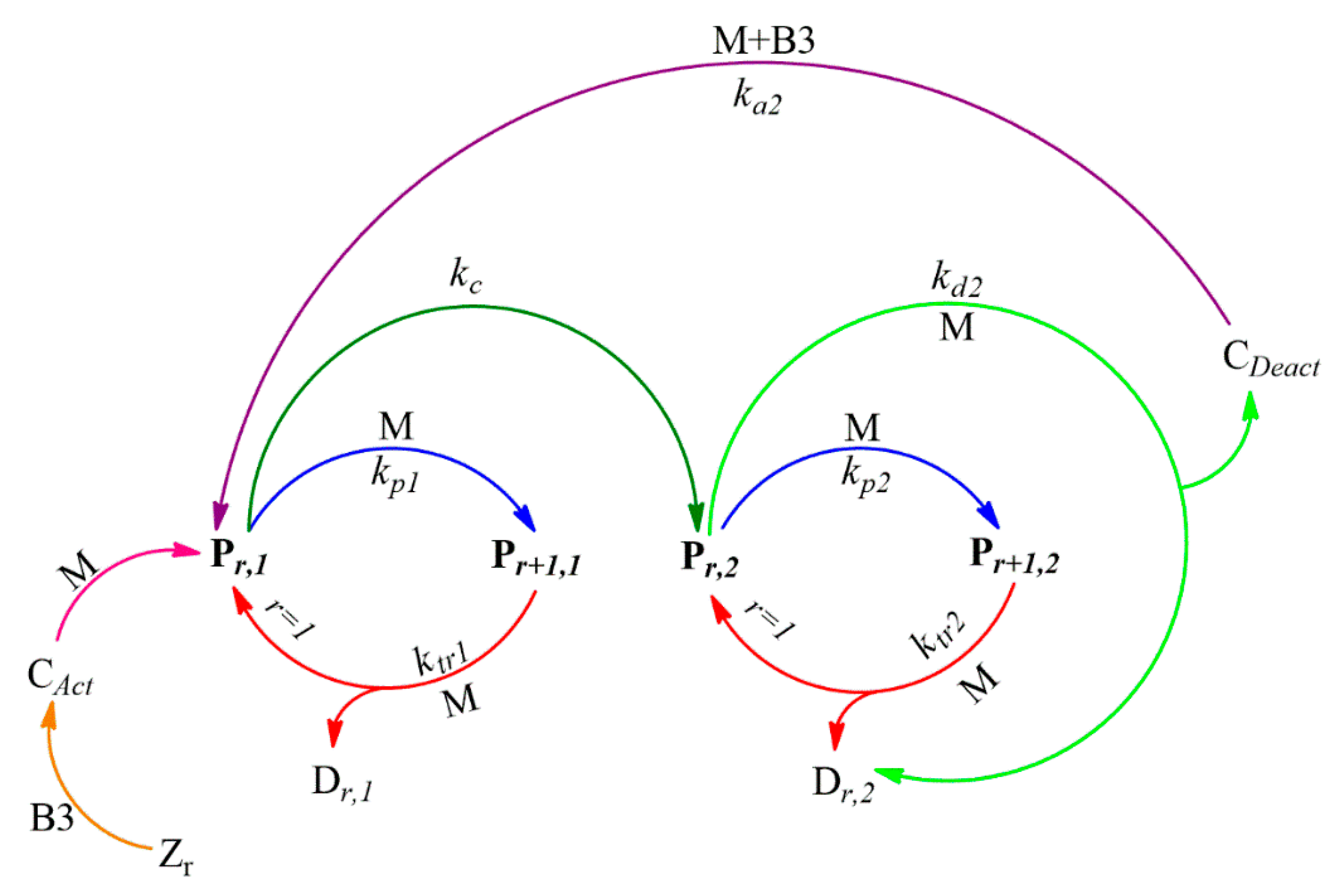
24] carried out a semi-batch reaction of a constrained geometry catalyst with MAO, and a kinetic model was proposed and then refined based on monomer uptake curves and polymer yield data. The deactivation of the catalyst/MAO system during ethylene polymerization was of the first order; the mechanism also included reversible activation and deactivation with MAO. The mechanism described the full kinetic picture. To the best knowledge of the authors, no study exists dealing with the modeling of zirconocene catalyst coordination polymerization using organoboron activators.
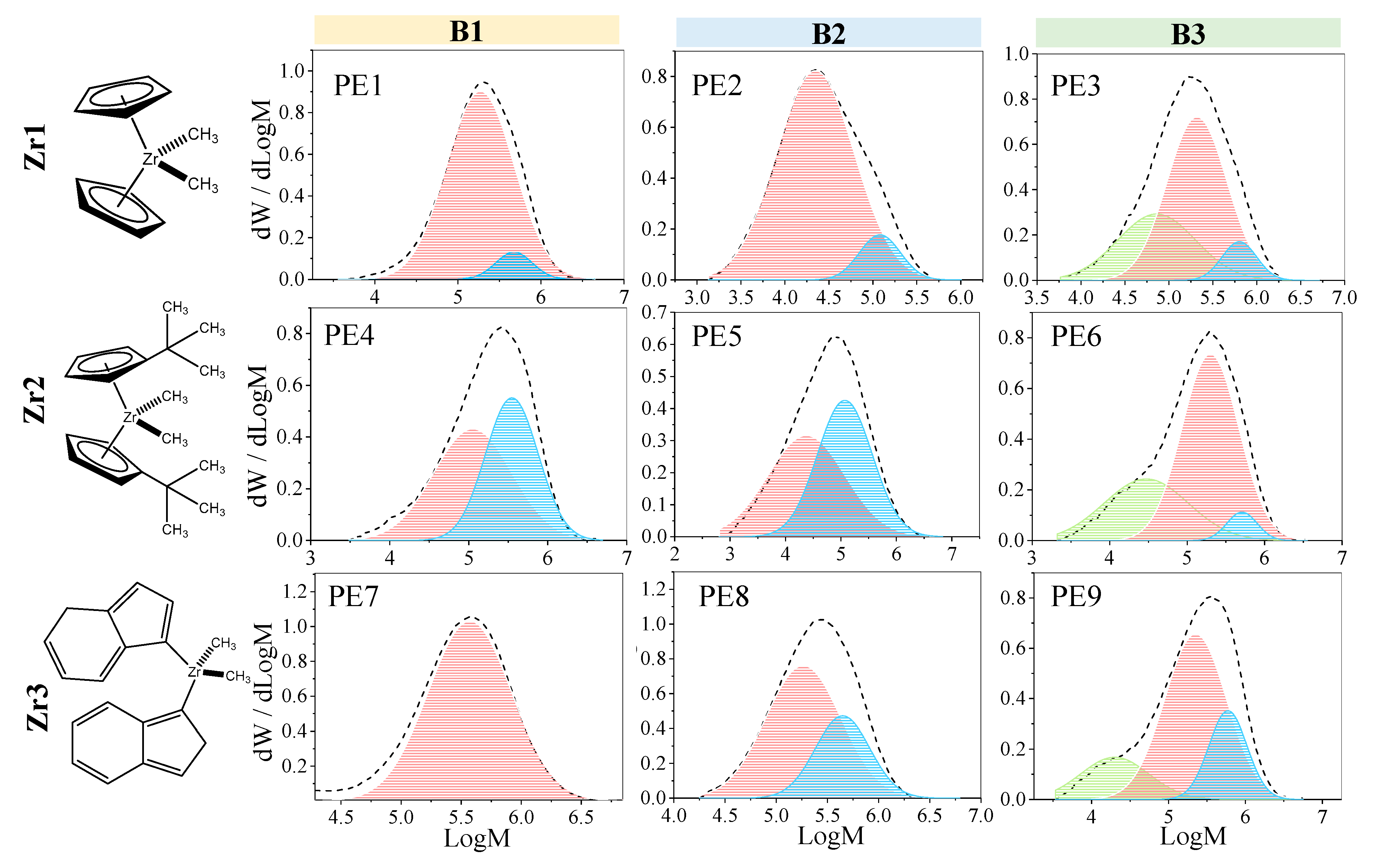
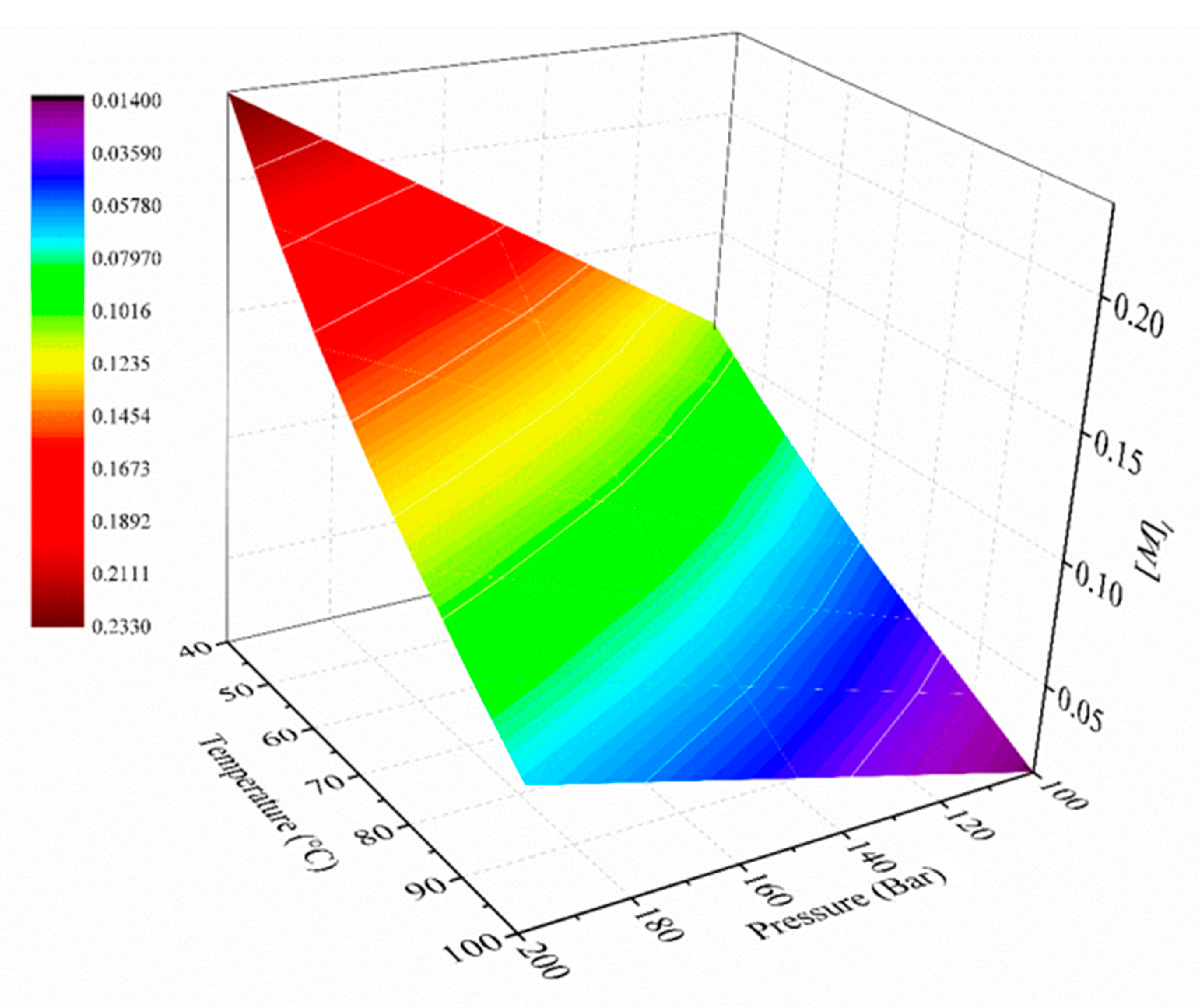
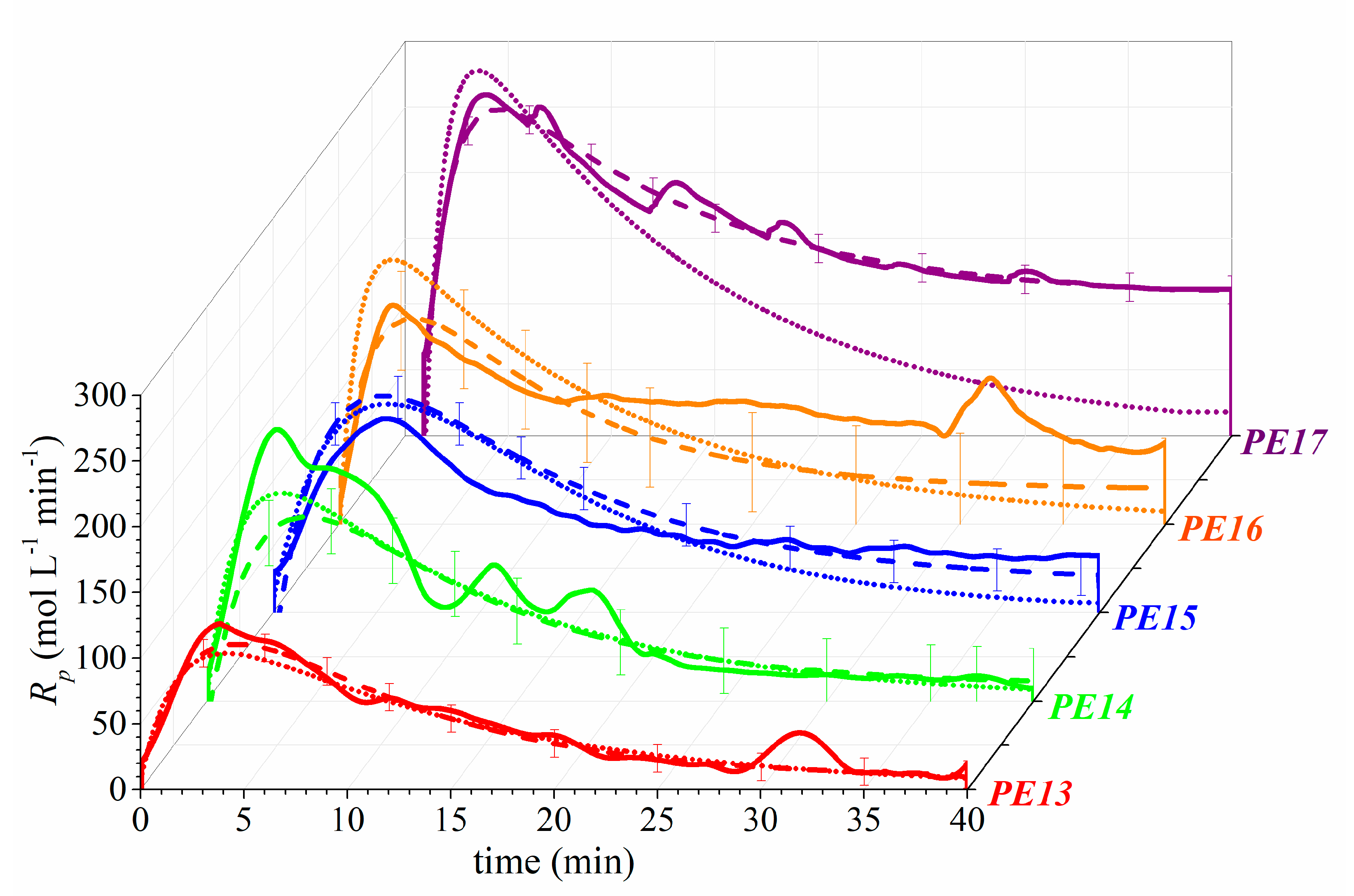
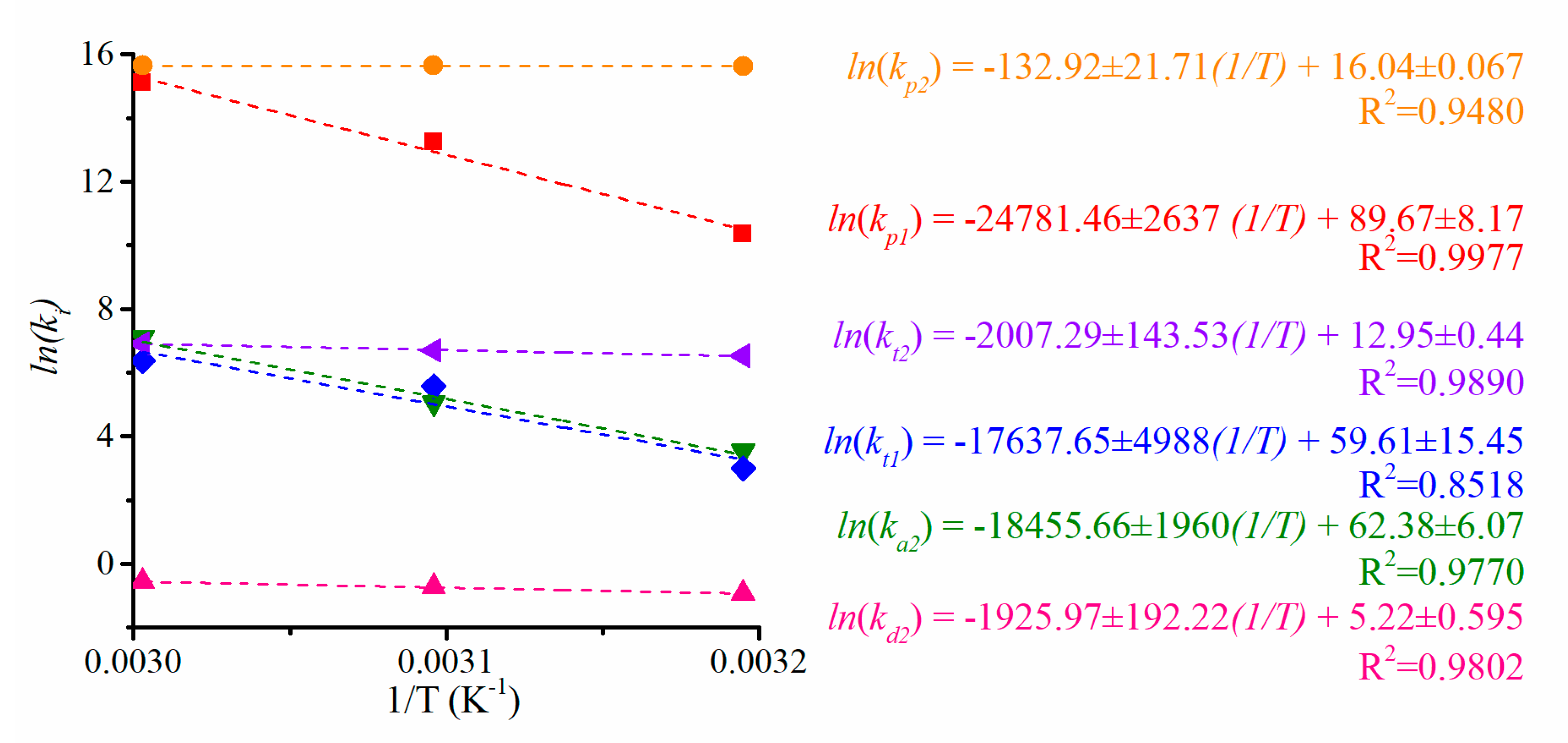
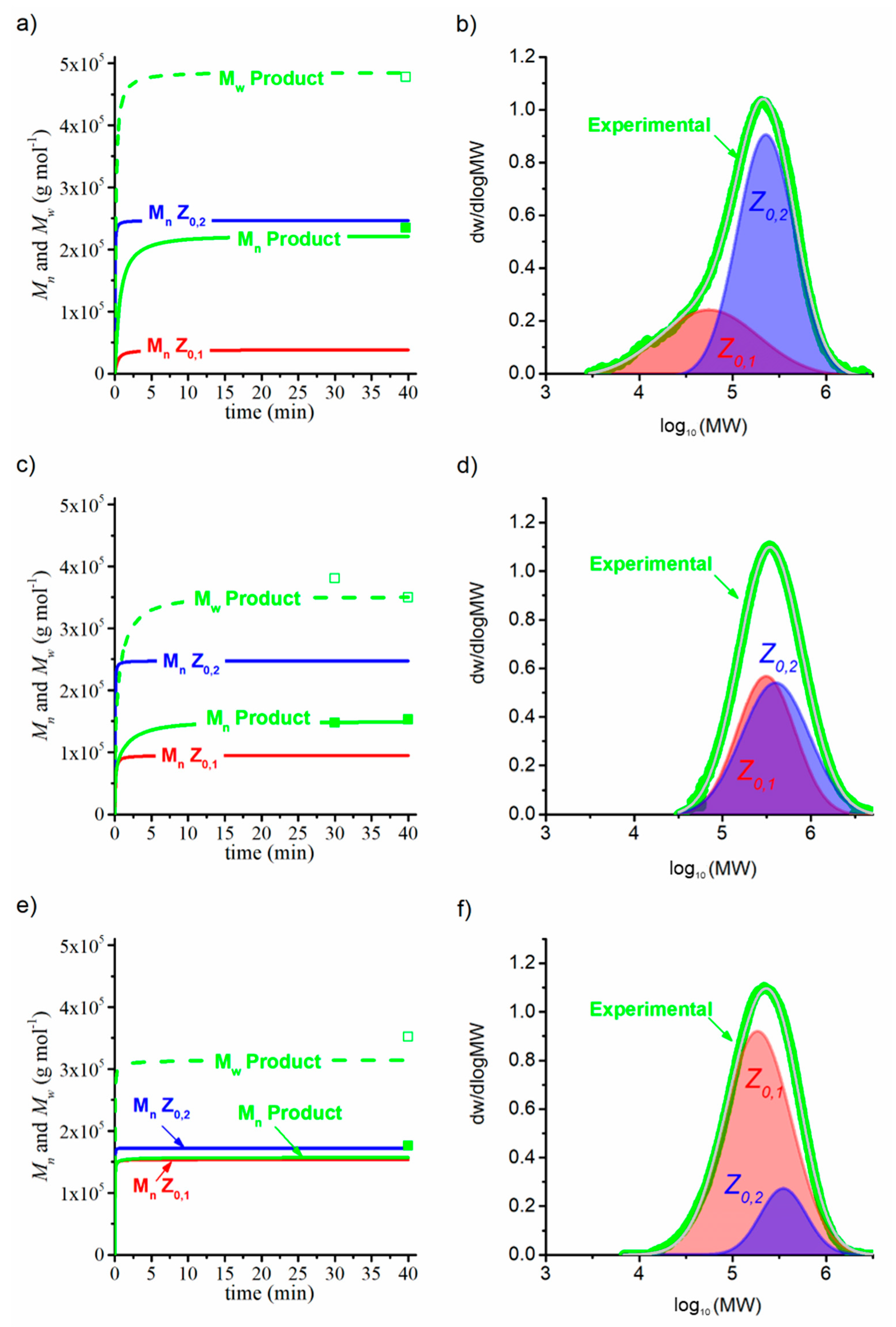
In this work, we aim to provide insights into the polymerization of ethylene catalyzed by Zr catalysts activated by organoboron compounds. Three Zr-based catalysts, with different ligand substituents, activated by three different organoboron compounds (B1, B2, and B3), were used for the PE synthesis. This work is focused on establishing the relationship between the catalytic system configuration with the polymerization behavior and with the final properties of the resultant polymers, in terms of molecular weight characteristics, crystallinity, and thermal behavior. Furthermore, the catalytic system leading to the highest catalytic activity was further analyzed, employing different solvents to elucidate the role over the features of the polymers. Moreover, a kinetic mechanism is proposed for the B3/Zr catalytic system, based on previous studies of MAO, and a mathematical model has been developed to estimate the kinetic rate coefficients of the two types of active species in the propagation, the chain transfer to monomer, polymer transition, spontaneous deactivation, and reactivation steps. With the knowledge of the kinetic parameters, the catalytic system is deeply studied, and some unexpected behaviors are analyzed.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}