1. Introduction
Proton transfer is one of the most ubiquitous elementary steps in many biochemical and biophysical processes. It is at the heart of acid–base-catalyzed reactions, before and after the actual bond-breaking or bond-forming step, or tautomerizations which transform a molecule in a related, yet different one with, e.g., different hydrogen bonding patterns. Proton transfer is also relevant in the context of electrolytic solutions, not the least because of the auto-protolysis of water molecules into hydroxide and hydronium ions.
Proton transport through membranes, via membrane proteins, is one such fundamental biochemical process. In most cases, such proton transport is highly regulated and often also coupled to a reaction that provides the energy for the “pumping” of protons against a chemical gradient. Famous examples of proton pumping membrane proteins belong to the respiratory chain: respirator complexes I and II and cytochrome c oxidase (complex IV).
Aside from protons being transported by a water molecule as a carrier, that is, a hydronium ion, an established mechanism of proton transfer in aqueous solution and through proteins, sometimes over larger distances, is the “proton relay” mechanism described by Grotthus [
1]. In this mechanism, a hydrogen-bonded chain is formed between a donor molecule, e.g., a hydronium ion, and an acceptor molecule, e.g., another water molecule, that can span several other water molecules. The proton transfer itself is then the excess proton moving from the donor molecule to its hydrogen-bonded neighbor, that molecule passing the proton on to its hydrogen-bonded neighbor molecule, etc. until the acceptor molecule receives a proton from its hydrogen-bonded predecessor. Similarly, a proton-hole can be transported by first the acceptor molecule taking a proton from its hydrogen-bonded neighbor, then that neighbor accepting a proton, etc.
Studying such processes by molecular simulations is a challenge because of the quantum mechanical nature of such a process. Even when neglecting the possibility of proton tunneling, which can be justified at physiological temperature and in biological systems, the change in electron density (and thus the associated atomic charges) together with numerous bonds being broken and formed more or less simultaneously, renders the application of classical force field approaches inadequate. Yet, such approaches allow molecular dynamics (MD) simulations to be performed at time scales which would be sufficient to observe spontaneous proton transfer events, if only the fixed topology (predefined bonds and fixed atomic charges) allowed such a transition. Several remedies have therefore been developed, all having their merits and difficulties. Here, we present only a few approaches.
Fixed-charge models can be overcome by hyperpolarizable force fields such as AMOEBA [
2], Drude models [
3,
4], or in combined quantum mechanical/molecular mechanical approaches. The latter suffer from the significantly higher computational cost, in particular if large parts of the system have to be described quantum mechanically. The way out here is to either use adaptive methods in which the partitioning of the QM and MM level is adjusted on the fly, see, e.g., in [
5,
6,
7,
8]. An alternative is to circumvent quantum mechanical calculations entirely and emulate the behavior, i.e., change of bonds and protonation states, as best as possible by classical calculations. This can be achieved by employing several predefined (protonation) states, and corresponding, typically QM-derived effective potentials, and switching between those as done in empirical valence bond approaches [
9,
10], reactive force fields [
11,
12] and multiscale reactive MD simulations based thereon [
13]. Other methods such as Q-hop [
14] involve a Monte Carlo step in which a proton transfer to an acceptor is proposed, based on varying criteria, and accepted by, e.g., a Metropolis criterium, i.e., evaluating the relative probabilities of the end states of the transfer in the desired thermodynamic ensemble. Before and after a proton transfer attempt, successful or not, the system is let to evolve freely for a period. The transfer itself can be a direct jump or combined with a
-dynamics in which the transferred proton is slowly grown and annihilated such as implemented in the HYDYN approach [
15].
Another combination of non-equilibrium MD and MC simulations is employed in constant-pH simulations [
16,
17,
18,
19] which allow the simultaneous change of protonation states at severaltitratable sites. In contrast to the other above mentioned methods, more than one proton is added or lost, as opposed to sampling the proton transfer probabilities, and they are mainly used to predict pKa values [
20]. Discrete protonation states replica-exchange MD simulations [
21,
22] follow a similar idea, that is, combining MD simulations with Monte Carlo steps for crossing between replicas, simulated at different pH and thus protonation states (instead of MD simulations at different temperatures as in the original replica-exchange MD). This approach allows estimating the probability for a proton to be located at a titratable site and thus its pKa value. Another view, in which only one excess proton is allowed in any of the replicas, can thus provide probabilities for the excess proton to sit at one or the other site and as such give an estimate of proton transfer probabilities between those sites. Such estimates can, as free energy differences, also be obtained by
-dynamics, but with different computational requirements, e.g., spending time in sampling artificial intermediate states at
while being free from the need to sample several transfer attempts.
In this work, we employ a simplistic variant of discrete protonation-state replica-exchange simulations to explore the proton transfer probabilities on the K-channel of cytochrome c oxidase (CcO). The K-channel has been discussed as the route for proton uptake in the situation in which the first reducing electron has arrived at the binuclear center (BNC), represented by the O→E intermediate state [
23,
24,
25,
26]. The channel is named after residue K362 (
Rh. sphaeroides numbering) and leads from a glutamate residue, E101, at the N-side of the membrane to residue Y288, located at the BNC [
27,
28]. Mutation of K362 [
29] showed significantly reduced transition rates to the next redox states in the catalytic cycle of CcO, suggesting that K362 is directly involved in the proton transport. Previous work has shown that the conformational dynamics of K362 depends on its protonation state [
30,
31], which in turn has been discussed to depend on the redox state of CcO, and that proton transfer is coupled to the conformational transitions of residue K362, and redistribution of water molecules and thus the hydrogen-bonded network [
32].
Here, we are interested not only in where the excess proton can be located, as specified by either protonated protein residues or positions of a hydronium ion, but also whether a proton transfer from and to such sites is possible. To this end, we perform several molecular dynamics simulations of CcO with one excess proton bound at various protonation sites. These are the titratable residues H96, E101, and K362 and the water molecules inside the K-channel. From those simulations in different protonation replicas, we evaluate at intervals the proton transfer probability as the exchange probability between replicas. Note that we do not carry out actual replica exchange simulations, and thus do not test whether a proton resides on the acceptor or could easily jump back. Rather, we evaluate which transfers are in principle possible. We therefore augmented the usual Metropolis criterion by a hydrogen-bond criterion. As in a Grotthus mechanism, the proton relay takes place along a hydrogen-bonded chain, proton transfer probabilities are highly dependent on the formation of such chains. Therefore, in our approach, a crossing attempt is only made to acceptor sites that are connected with the proton donor by a hydrogen-bonded connection. We find that this additional criterion severely reduces the number of accepted proton transfer attempts. The surviving proton transfers, however, provide valuable information about possible routesfor proton translocation in the K-channel of CcO.
3. Results and Discussion
Figure 1 shows two snapshots taken from the MD simulation of the full model. In one snapshot (
Figure 1a), protein residue K362 points towards the upper part of the K-channel with torsion angles
,
,
, and
. This conformation will be called “up”. The other, “down”, conformation (
Figure 1b) has torsion angles
,
,
, and
. Because the two outermost torsion angles
and
are far from linear, this conformation does not point fully down to the channel entrance, but rather in direction of residue S299 (see
Figure 1b). In each of the two snapshots, there are fifteen water molecules inside the K-channel which are considered as possible carriers of the excess proton (instead of K362). In the “up” model the height of the K-channel as defined by the distance between the CA atoms of H96 and Y288 is 25.2 Å, whereas in the “down” model, this height amounts to 27.4 Å. With respect to the CA atom of K362, Y288 is located ∼1 Å higher and H96 is located ∼1 Å lower in the “down” models than in the “up” models.
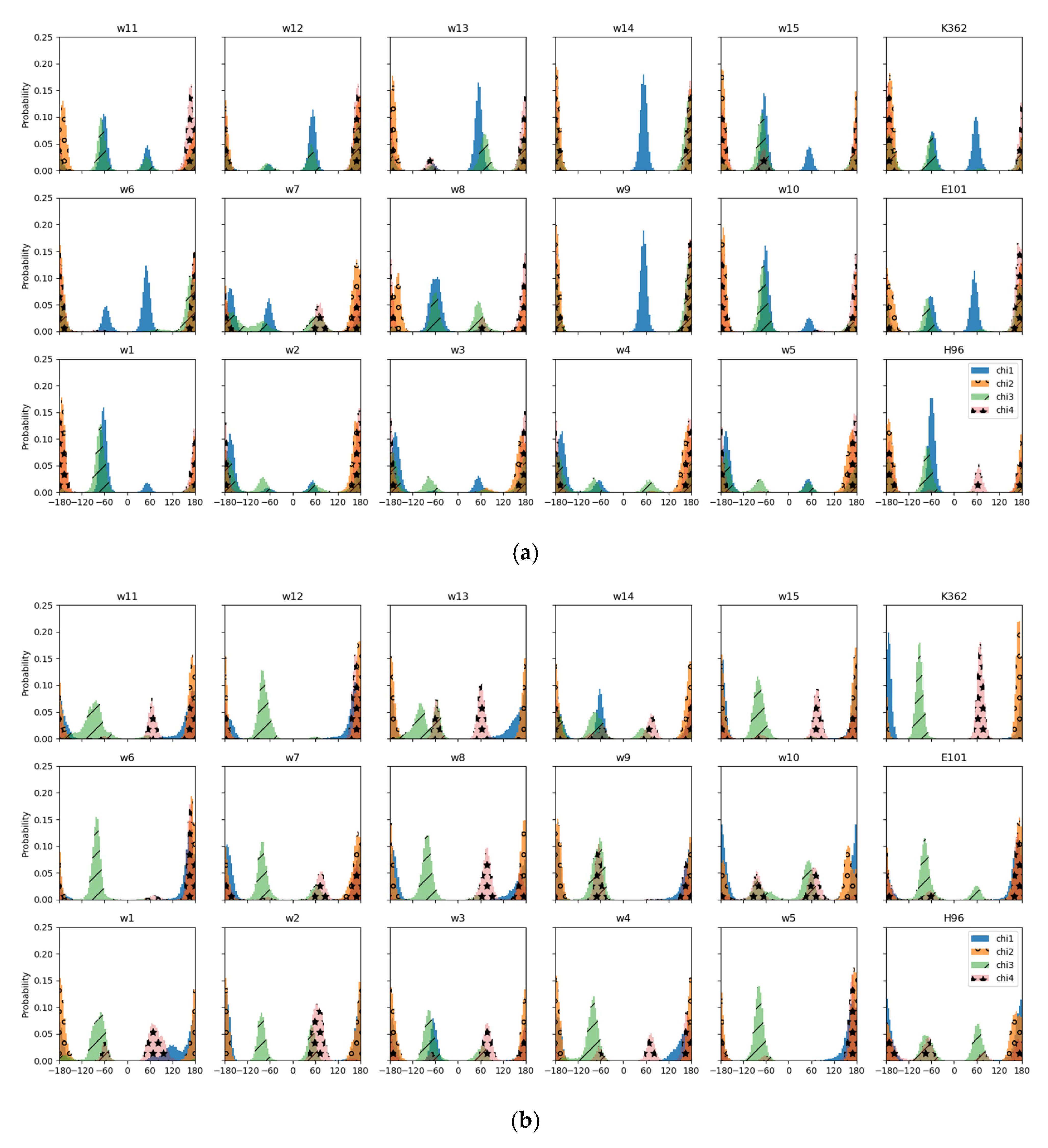
3.1. Conformational Analysis
3.1.1. Protein Residues
Of the protein residues, only K362 shows some dependence of the average position of its polar side chain atom, NZ, on the protonation model, that is the location of the excess proton. In the “up” models it fluctuates up to ∼3 Å, and in the “down” models ∼2 Å around a height of 17 Å (see
Figure 2 and
Table S1). For the other residues, the positions of their polar side chain atoms remain rather stable, some of them with different positions in the “up” and “down” models, partially due to the shifted reference point, the CA atom of H96: the hydroxyl oxygen atom of Y288 is located at ∼24 Å in the “up” models and at ∼26 Å in the “down” models and the CD atom of H96 fluctuates around 2.9 Å and 0.8 Å in the “up” and “down” models, respectively (see
Figure 2 and
Table S1).
In the simulations of the reduced “up” model, in which also K362 is protonated, the torsion angles are in the same state as in the full model, except for
which is a deviation from the almost-linear conformation of the “up” snapshot (see
Figure 3a). Indeed, in the other “up” models, angle
varies between almost linear and
, without any clear dependence on the location of the excess proton. A conformation of
can also be observed in all protonation models based on the “down” snapshot, albeit with varying intensity. Torsion angle
is about linear in all models.
The difference in torsion angle of nonlinear, , in the “up” and linear in the “down” conformation of the full model is preserved in almost all the reduced models. Exceptions are the “up” models w2, w3, w4, and w5 which exhibit a linear angle.
Among the models based on the “down” snapshot, torsion angle
varies remarkably. This angle is about linear in models w5, w6, w12, and in the model with protonated E101 and
for the other “down” models (see
Figure 3b). It is also about linear in the “up” models (except for model w7 and that with protonated H96).
In contrast to the “up” snapshot of the full model, exhibits about linear conformation in some of the reduced models, namely, models w2, w3, w4, and w5.
Residue E101 exhibits a slightly shifted
angle only when it is protonated itself and then only in the model based on the “up” snapshot (see
Figure S1), but is otherwise unaffected by the protonation model. The conformation of residue H96 is independent of the protonation model of either protein residues or water molecules but different in the “up” models and “down” by ∼
in
and ∼
in
(see
Figure S2). This difference in H96 conformation together with the altered relative heights of H96 and E101, results in a shorter distance to E101, allowing a hydrogen bond between the two residues.
3.1.2. Water Residues
The fifteen water molecules, as illustrated by
Figure 2, show a much higher mobility in the “up” models than in the “down” models. It is interesting to note that in the “up” models there is a region at about 13–14 Å, a height between S365 and K362, which is almost unoccupied if the proton is located at a water molecule above that zone or at K362.
In both types of models, some water molecules are more mobile than others, though. The average positions of the water molecules are given in
Table 1.
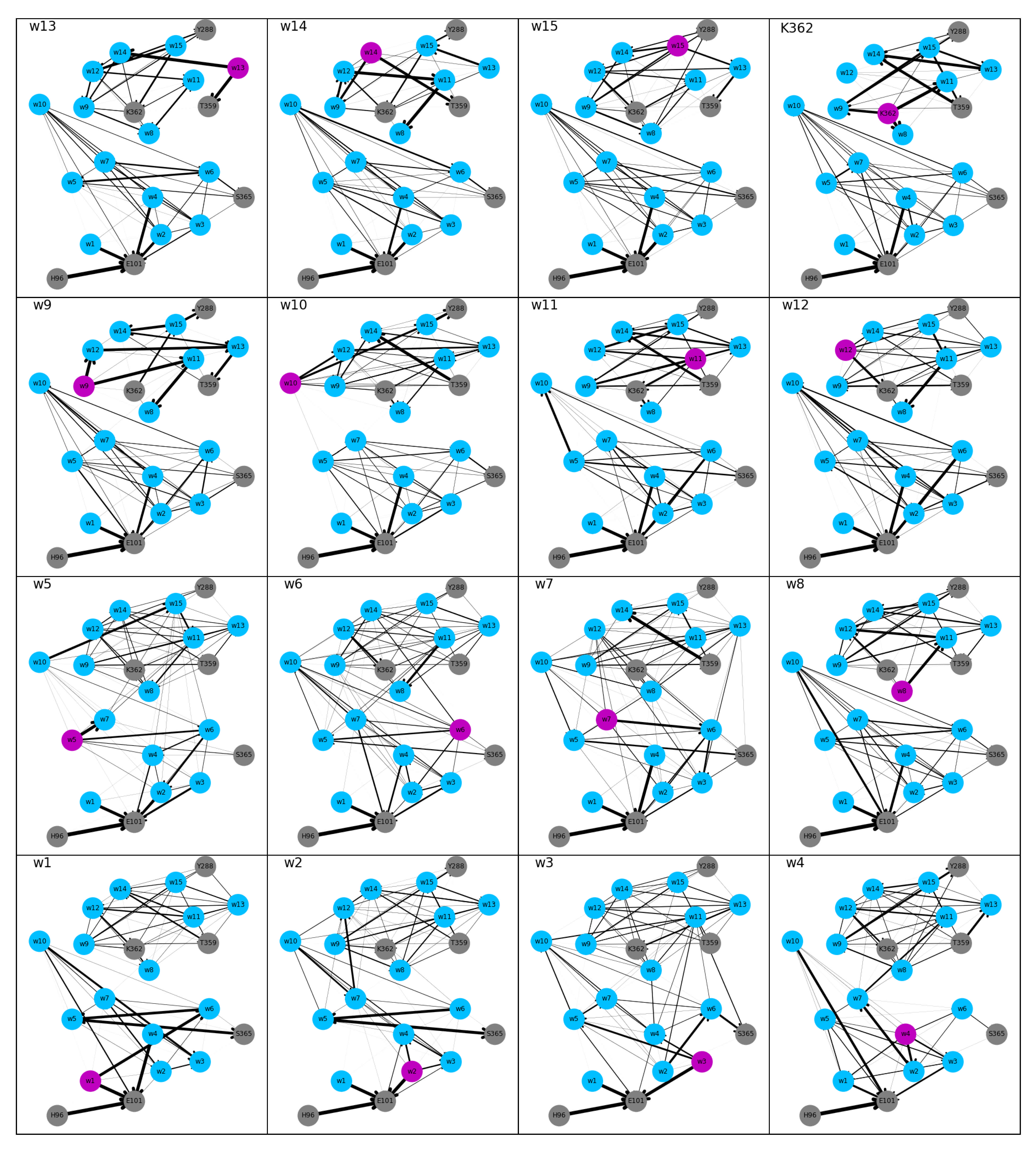
3.2. Hydrogen Bonds
Most of the undirected hydrogen-bonded networks in the “up” models are partitioned in two, at about the height of residue K362 or just below, i.e., at the height of w8 (see
Figure 4), due to no or very low probability (models w9 and w10) for hydrogen bonds between water molecules in the lower and the upper part of the channel. This gap in the networks can be related to the low probability for water molecules to be at such a height (cf.
Figure 2). Networks in which all nodes share one connected component are those of models w2, w3, w4, and w6. In model w7, the upper and lower parts of the network are connected, but the network does not reach Y288. Connections within the upper part of the network are dense in all “up” models. The lower part, however, shows lower connectivity for models w1–w7, that is, models in which the proton is located mainly in the lower part. For directed hydrogen-bonded networks of the “up” models, the probability for connections is substantially reduced compared to the undirected models (thinner lines in
Figure 5 than in
Figure 4).
In the “down” models, Y288 is not connected to the rest of the hydrogen-bonded network or only with very low probability, in agreement with the low probability to observe a water molecule just below this residue (see
Figure 2). All other residues are connected in both the undirected and the directed networks of the “down” model, albeit some with low probability (see
Figure 6 and
Figure 7, respectively). The connectivity, in terms of number of edges, of the undirected networks in the “down” models is generally lower than in the “up” models, in particular in the upper part of the networks (see
Figure 4 and
Figure 6, respectively).
In all “up” models, a strong hydrogen bond between H96 and E101 exists that is not formed in the “down” models. The direction of the “up” hydrogen-bonded network is, in the lower parts, generally towards E101, whereas in the “down” models hydrogen bonds point in all directions although E101 is always an acceptor, except when it is itself protonated and acts as donor and acceptor.
For the proton to reach the upper part of the network along directed hydrogen bonds, it has to be located on a water molecule that has an average height of >8 Å (w4) in the “down” models, in the “up” models a height of >16 Å (w8) is required for directed hydrogen-bond connections between the proton-carrying residue and the upper part of the network. Once the proton has reached this height, however, no directed hydrogen-bond connections to the lower part (with probability higher than 0.3) exist, preventing the back transfer. This is again in contrast to the “down” models, which also show hydrogen-bonded connections towards the lower part of the network (and thus the K-channel) when the proton is located at or above K362. Indeed, K362 itself is accepting hydrogen bonds mainly from the part of the channel where the proton is located, that is, from below or above. In the “up” models, K362 almost exclusively accepts hydrogen bonds from above, even when the proton is located in the lower part of the channel.
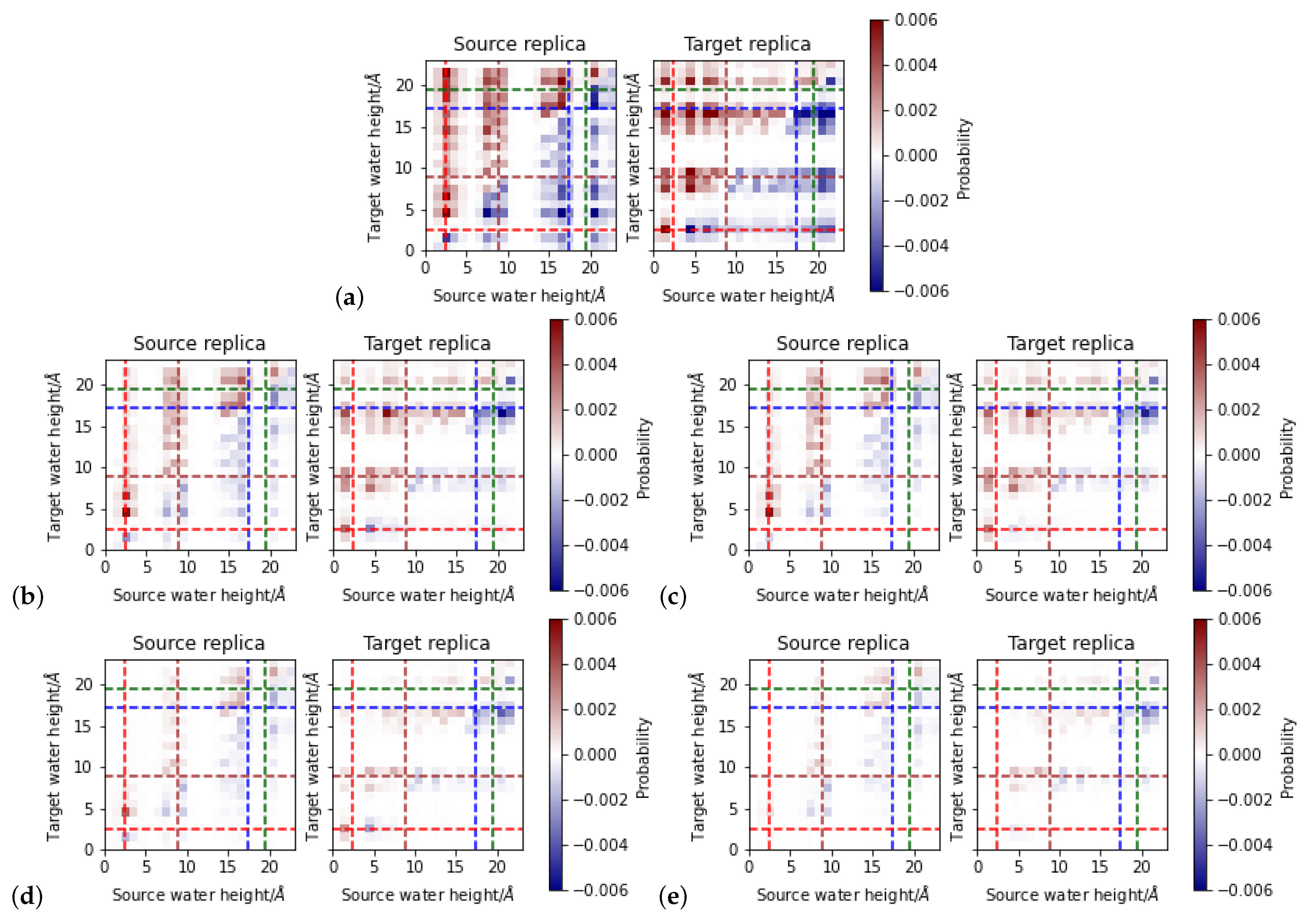
3.3. Proton Transfer Probabilities
The acceptance ratio of proton transfer without any of the hydrogen-bond criteria is about 0.5. An exchange is attempted in both directions, “downwards” or “upwards”, and the ratio of about half of the attempts accepted suggests that the direction, and thus the energetically preferred location of the excess proton, is dominant. Given the large energy differences between individual frames (up to almost 100 kcal/mol, see
Figure S4),the acceptance probability for a transition into a highly unfavorable position can be expected to be rather low. On the other hand, for all heights at which protonated water molecules are located, such a large energy range is observed. Consequently, there will always be a transition attempt from one height to another that is energetically favorable and as such accepted for sure but the reverse transition is only accepted with very low probability, explaining the about one half acceptance ratio.
Upon addition of a hydrogen-bond criterion, the acceptance ratio drops to ∼0.2 when requesting hydrogen bonds only in the source replica and further to 0.09 when requesting hydrogen bonds in both the source and target replica. The further constrained of directed hydrogen bonds reduces the acceptance ratio to ∼ and to ∼ for hydrogen-bonded connections in only the source and in both replicas, respectively. This suggests that most of the relevant hydrogen-bonded connections along which proton transfer is energetically allowed (with a certain probability) are anyway directed. The source water molecule, as hydronium ion, must be a hydrogen bond donor. The analysis of the directed-hydrogen bond networks shows that, at least in the “down” models, and in the lower part of the “up” models, the excess proton has a directing effect on the network.
3.3.1. Height-Dependent Proton Transfer Probabilities
Figure 8 and
Figure 9 summarize the position-dependent acceptance for proton exchange between the “up” and the “down” models, respectively, depending on the different criteria. For both types of models, there is a zone between 10 and 14 Å from or to which no proton transfer is accepted. This is the zone which is not occupied by any water molecule in those “up” models which have the proton located in the upper part of the channel. In the “down” models, water molecules can be observed in that zone, but hardly a protonated one. Accordingly, no proton transfer from this zone is attempted and as such also not observed.
For the “up” models, the proton transfers not accepted due to the additional hydrogen-bond criteria are all “downwards”, starting from a height between 16 and 20 Å in the channel, mainly between the height of the NZ atom of K362 and the OG atom of T359. These differences show that, while protonated water molecules can occupy that region (and can exchange protons if only energetics are considered), they are not part of a hydrogen-bonded network that leads to a target water molecule. This is consistent with the observation of “disrupted” networks, ending at the height of K362 in the “up” models (
Figure 5). The additional requirement of a hydrogen-bonded connection also in the target replica further reduces the accepted attempts by those proton transfers which would reach up into again that region and are also not possible due to the lack of a directed hydrogen-bonded connection.
The area between K362 and T359 at ∼13–14 Å exhibits a striking difference between the “up” and “down” models. In the former, the highest probability for proton transfer is from and to the regions between K362 and T359, whereas in the “down” models, proton transfers can only land there, although protonated water molecules are observed at this height in the MD simulations of both “up” and “down” models. This suggests that in the “down” models a height between K362 and T359 is energetically much more favorable for the excess proton than any other possible target water molecule location in the channel. Relating this observation with the higher fluctuations of the conformations of K362 and T359, and thus also heights of their polar side chain atoms, the protonated water molecule may be more tightly accommodated between the two residues in the “down” models. In models w9, w10, and w11, the protonated water molecule is found at about the height of K362 and it is also hydrogen-bonded to this residue.
Another difference between the “up” and “down” models originates in the conformations of H96 and E101 at the bottom of the channel. In the “down” models, only very few proton transitions start at a height of ∼5 Å. With additional hydrogen-bonded connections as further prerequisite for a possible proton transfer, this ∼5 Å zone becomes void of accepted proton transfers. This area is occupied by a hydronium ion only in the simulation of model w3 and in this model, the hydronium ion is rather mobile. Consequently, proton transfers into this zone are accepted, rendering the energies of the respective snapshots sufficiently favorable. This observation suggests a position at a height of ∼5 Å can in principle be occupied in the “down” models, but rather transiently, as shown by the low probability for doing so in the MD simulations. A possible explanation is the vicinity of the negatively charged E101. In an MD simulation, this residue would significantly attract a protonated water molecule, leading to the formation of strong hydrogen bonds and rendering a position of the hydronium ion farther up the channel much less favorable.
In the “up” models, there are transitions observed from and to a height of ∼5 Å, mainly downwards, and thus also likely influenced by the negative charge of E101. Still, for transitions to start at ∼5 Å, in those models hydronium ions have to occupy this area also in the MD simulations. In the “up” models, the lowest average height of the protonated water molecules is at Å (w1, w2, and w3), suggesting that the presence of E101 is less attractive in those “up” models than in the “down” models. This in turn can be explained by E101 forming a strong hydrogen bond with H96 in the “up” models, which does not exist in the “down” models and provides some shielding of E101’s negative charge.
3.3.2. Directionality and Lengths of Proton Transfer Paths
For the “down” models, proton transfer starting below K362 is only upwards, consistent with the direction of the hydrogen-bonded network. The longest paths are found for source water molecules located at the bottom of the channel. Paths starting from positions higher up in the channel are shorter, indicating that there are no “detours” along the hydrogen-bonded chains. At just below K362, some longer paths leading down the channel are also observed. Moreover, proton transfer paths starting above K362 all lead “downwards”, again in agreementwith the directionality of the hydrogen-bonded network. Paths spanning from the bottom of the channel to above K362, or vice versa, reach lengths of more than 10 residues involved (see
Figure 10).
The “up” models show proton transfer starting from about the height of T359 and above is mainly in “downwards” direction, whereas paths starting just below K362 lead “upwards” (see
Figure 11). This is also reflecting the directed hydrogen-bonded networks which point “down” from the protonated water molecules in models w13, w14, and w15 in the top part of the channel, and mainly up in models w9 and w10. If only the hydrogen-bonded connections in the source replicas are considered, paths starting at the lower end of the channel do reach up to the same lengths in the “up” models as observed in the “down” models, even though the protonated water molecules in those cases are located significantly more above E101 than in the “down” models. Implementing the stricter criterion of a hydrogen-bonded connection also in the target replica reduces the probability for long paths significantly in the “up” models. The paths starting above K362 reach lengths of six residues as opposed to more than 10 residues observed for the “down” models, in agreement with the disrupted hydrogen-bonded networks in the “up” models.
It is also interesting to note that, while paths starting at about the height of S365 can have either direction in the “up” models, as long as only the hydrogen-bonded connection in the source replica is relevant, only the “downwards” paths survive the test for the additional hydrogen-bonded connection in the target replica. In the “down” models, in contrast, paths starting up to the height of S365 always lead “upwards”. In these “down” models, the stricter criterion reduces the overall probability but does not affect the predominant direction. In these models, the “fork in the road” of the more probable direction of proton transfer is at about the height of K362.
The stricter criterion of directed hydrogen-bonded connections, either in the source replica only or in source and target replica, reduces, as anticipated, the probability for proton transfer in “up” and “down” models, evenly to and from all regions that show proton transfer along undirected hydrogen bonds.
3.4. Interplay of K362 Conformation, Hydrogen-Bonded Connections, and Proton Transfer Probabilities
The overall proton transfer acceptance probabilities of about one half based on energetic considerations suggest that the energy difference between the source and target replica, and thus also the position of the excess proton, is dominant. Further consideration of hydrogen-bonded connections, by analyzing the hydrogen-bonded networks, and by introducing the existence of such a connection as an additional criterion for the acceptance of a proton transfer, afforded us with an understanding of preferred proton transfer directions (“upwards” or “downwards”) and its dependence on the location of the excess proton. The differences between the models built from a snapshot with K362 in an “up” conformation and those built from a snapshot with K362 in a “down” conformation, gave us furthermore insight into the interplay of the hydrogen-bonded connections with the conformation of this residue, and thus also its possible role in proton transfer through the K-channel of CcO. In the “down” models, proton transfer is possible throughout the channel, that is, from the lower part up to almost Y288 or in the other direction, depending on the position of the excess proton. In the “up” models, in contrast, the hydrogen-bonded networks in the channel are disrupted such that lower and upper part of the channel, below or above K362, are disconnected. Consequently, proton transfer along hydrogen-bonded chains is only possible within the lower or upper part. Once in the upper region, hydrogen-bonded connections between the proton carrier and the terminal residue of the channel, Y288 exist, and a transfer to this residue is conceivable. Passage from lower to upper part has thus to take place while K362 is in a “down” conformation. Whether it is K362 itself that carries the proton up or not has not been considered in this work. Extensive QM/MM simulations [
32] show that proton transfer from K362 to Y288 is a feasible pathway and facilitated by an “up” conformation of K362. Our data suggest that the proton transfer can also occur between water molecules as long as K362 remains “down”. The “up” conformation has been discussed to be predominant for protonated K362 but may also have higher probability for an excess proton in the vicinity of K362, that is, in the upper part of the channel. Among our “up” models, those with a proton in higher positions indeed show a preference for an “up” conformation of K362, whereas models with an excess proton located towards the bottom of the channel also exhibit “down” conformations. In the “down” models, K362 does not flip up, indicating that in the rather short MD simulations used to sample the water positions and hydrogen bonded connections such a transition is not feasible. In longer simulations [
30,
33], such transitions are observed, rendering proton transport by K362 as a carrier a possible, but not the only route. Instead, K362’s role can be regarded as a valve, separating the upper and lower parts of the K-channel and, in an “up” position, preventing proton transport back to the bottom of the channel but possibly also the translocation of another proton before the next step in the catalytic cycle. In [
32] it is discussed that “The side chain dynamics of the neutral Lys-319 and the consequent loss of a hydrogen-bonded connectivity could help in preventing the chemical proton from leaking backwards towards the N-side.” The “loss of hydrogen-bonded connectivity” [
32], is not observed in our “down” models, as it is also a consequence of the significantly lower channel hydration with neutral lysine, and we have kept the amount of water molecules in the channel constant at fifteen to facilitate comparison between our “up” and “down” models.
3.5. Strengths and Limitations of the Presented Sampling Approach
Our present sampling approach of proton transfer probabilities along hydrogen-bonded chains is not a true replica exchange approach as the simulations are not continued after exchanges and therefore possible direct jumps back are artificially excluded. This can be seen by proton transfers that are accepted into unfavorable regions for hydronium ions but not out of regions unfavorable for hydronium ions. While such a behavior seems to violate detailed balance, it is mainly a problem of too little sampling of the source distribution: the conditional probability to transfer from an unfavorable proton position to a more favorable one (which will for sure be accepted) vanishes because the condition for the proton to be in that position is very low according to the MD simulations. Continuation of the MD simulations after exchanges, as is done in actual replica-exchange simulations, is a straightforward way to improve the sampling of those otherwise difficult to reach states. We have in this work instead focused on the effect of the hydrogen-bond criterion in the acceptance step. The request for an existing hydrogen-bonded connection reduces the accepted proton transfers to those cases in which a proton-relay mechanism is conceivable. By allowing hydrogen-bonded connections of any length, this approach also allows proton transfers over significantly larger distances than hopping to only the next hydrogen-bonded neighbor and thus also includes the possibilities of concerted proton transfer events.
For simplicity and to be able to use standard empirical force fields for a direct comparison of energies, we have limited the sampling to proton transfer between water molecules. However, our approach can easily be extended to include protein residues. This, however, needs a QM/MM setting for the energy evaluation (thought not for the MD simulations) or other effective potentials that allow the comparison of different topologies such as provided by EVB [
9,
10] or reactive force fields [
11,
12].
As a validation for the proton transfer estimates provided by our method some further simulations with increasing complexity can be carried out. First, an actual, though artificialproton exchange could be carried out by placing the excess proton at the target water molecule of the target replicafollowed by a short MD simulation in this new configuration. This way, the probability for the newly generated hydronium ion to actually be found at the position of the target water molecules can be evaluated. As another, computationally more demanding, approachthe proton can be “transferred” by -dynamics, annihilating it at the source water molecule while creating an excess proton at the target water molecule.This way, free energy differences between the two protonation states can be computed. Moreover, finally, the actual proton transfer can be calculated by, e.g., umbrella sampling simulations, so as to obtain a free energy barrier for the proton transfer step as well as the free energy difference between the end states. For those calculations, however, a potential that can describe different topologies has to be used. Given the computational demand of these more elaborate methods, our simplistic approach can serve as a first, broad screening of which proton transfer steps may be feasible and worth inspecting with higher-level methods.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}