1. Introduction
Electrochemical applications have undergone a remarkable increase in importance during the last decades due to the need for efficient modern technologies, e.g., in syntheses of substances or mobility, but also due to the rapidly evolving availability of new experimental and numerical tools for precise study and characterization of electrochemical systems [
1]. One example of such an electrochemical device is the fuel cell (FC) for generation of electrical energy from oxidation of hydrogen or short-chained hydrocarbons like ethanol or methanol [
2,
3]. The most promising candidate for the large-scale production of hydrogen for use in fuel cells is the electrolyzer for water electrolysis [
4], which is itself based on electrochemistry. Moreover, batteries are currently facing strong interest from researchers, since they are an essential component, e.g., in modern microelectronic devices, in electromobility and in energy storage. Latz and Zausch gave thermodynamically consistent theoretical descriptions of transport phenomena [
5], reaction kinetics [
6] and thermal aspects [
7] in these devices. Another application of electrolysis is the conversion of carbon dioxide (CO
) [
8] to valuable products such as organic acids or short- and long-chained alcohols in order to reduce the global carbon footprint. A work realizing such a synthesis route experimentally to produce ethylene using copper(I) oxide catalysts can be found in [
9].
The process introduced in this work comprises the oxygen reduction reaction (ORR) mechanism, with hydrogen peroxide (H
O
) as the desired product. This important commodity chemical has a wide range of applicability in decentralized or industrial processes. In the chemical industry, it is synthesized via the anthraquinone process, delivering product purities of 1–2 wt%, which can be enhanced up to 70 wt% by downstream processing [
10]. Due to its high oxidation potential, it is commonly used for treatment of waste water or bleaching of paper pulp or textiles [
11,
12]. Furthermore, it is employed extensively in the production of fine chemicals [
13]. In the encymatic halogenation of the phenolic monoterpenes thymol and carvacrol to different products for anti-inflammatory, anti-microbial and anti-cancer applications, H
O
accompanied by the enzyme chloroperoxidase is used as a bio-catalyst [
14]. Getrey et al. showed that the highest conversion rates are reached with electrochemical in-situ synthesis of H
O
directly in the active zone for the following reaction [
15]. Thus, there are no transport problems for the catalyst as in the case of external injection of hydrogen peroxide. Since oxygen is used here as a reactant to produce H
O
, maximum efficiencies are achieved using gas diffusion electrodes [
16] to avoid the problem of limited solubility of O
in the electrolyte.
Decentralized electrochemical production of H
O
has received growing interest from researchers in recent years. Yang et al. perceive this as part of a general trend [
17]. They predict that the increased availability and decreased cost of renewable electricity will transform the chemical industry towards on-site production. This could lead to logistic challenges, e.g., if larger amounts of product have to be gathered. On the other hand, the decentralized approach has the potential advantages of more efficient energy conversion, easier product storage and lower capital expenditure according to Sehrish et al. [
18]. In this respect, Li et al. recently stated that gas diffusion electrodes, such as the one used in the current work (see
Section 2.3), are crucial to exploit the advantages of an electrochemical production of H
O
[
19]. They quantitatively assessed their reactor with respect to energy consumption, production capacity, cost and long-term stability. They conclude that the technology is already feasible for on-site abatement of pharmaceutical residues in ground water. As for long-term stability, they report a life time of their electrode limited to about 46 days due to flooding of pores. In a real-world application, the electrodes need to be replaced before expiration to ensure a site-independent product quality.
Inspiration for modeling electrochemical reactors can be obtained from theoretical works on fuel cell research, the quantity of which is higher than that for electrolyzers. Since FCs convert substances into electric power, they can be considered as the inverse operation of electrosynthesis. Schultz et al. set up a rigorous, one-dimensional model of a direct methanol fuel cell using a Flory-Huggins activity model [
20]. Later, the same group investigated the dynamic response of the system with respect to reaction kinetics [
21]. The basic features to be covered by a model in any case are mass balances, material conversion, potential drops and membrane flows. In the case of spatial resolution (dim. > 0), local transport effects need to be described, too. They may be Knudsen diffusion or Maxwell–Stefan diffusion, the latter being governed by molecular friction in porous media [
22], which is taken into account in [
20]. Furthermore, Weinzierl and Krewer used numerical simulations to analyze the water management in direct methanol fuel cells [
23,
24]. For a more sophisticated approach, see e.g., [
25], where a version of the 2D Navier–Stokes equations is solved for a mixture in order to investigate its coupling to chemical surface reactions via boundary conditions.
A mathematical model of an electrochemical cell for copper electrolysis, composed of 0D and 2D submodels, was given by Pohjoranta et al. [
26], who validated their theoretical predictions by comparison to cyclic voltammetry measurements. However, this work is concerned with the overall process. Details about unit operations are secondary as long as their main dependencies are captured with reasonable accuracy. Hence, we stick to a 0D reactor model such as the one of Görgün et al. [
27], who published a balance-based mathematical description of an electrolyzer with a proton exchange membrane (PEM), or the one of Argyropoulos et al. [
28], who set up a semi-empirical 0D model of a direct methanol FC in a similar fashion. The aspired philosophy of the model equations is to aim for fast convergence while realistically reproducing measured data for the most important output quantities, i.e., the product weight fraction and the Faraday efficiency. This is additionally beneficial, because it does not imply a high number of transport coefficients as in the case of spatial resolution. Such coefficients (e.g., the binary Maxwell–Stefan diffusional coefficients) are often unknown in praxi due to the limited availability of experimental data for parameter estimation. However, it will be necessary to thoroughly determine the kinetics of the reaction mechanism under consideration.
A means of studying electrochemical reaction kinetics, which relies on experimental data, is the rotating ring-disk electrode (RRDE) system [
29,
30]. The method is based on the rotation of circular electrodes, inducing a forced flow to drag reactants to the electrode. Then, the detected electric currents at the disk and at the ring, with the latter being located at higher radii, contain information about the reaction kinetics. Yet, a micro-scale simulation is required in order to extract this information from experimental results. An application to determine electro-kinetic rate constants of the ORR mechanism at gold catalysts can be found in [
31]. Recent reviews about catalysts for electrosynthesis of H
O
by oxidation of water can be found in [
32,
33]. Recently, two-dimensional RRDE simulations based on a finite-difference discretization were successfully adapted to corresponding measurements in a detailed and quantitative electro-kinetic study [
34]. Such a way of determining parameters of a unit operation by simulations on a smaller scale can be considered as a part of a bottom-up modeling approach, as sketched in
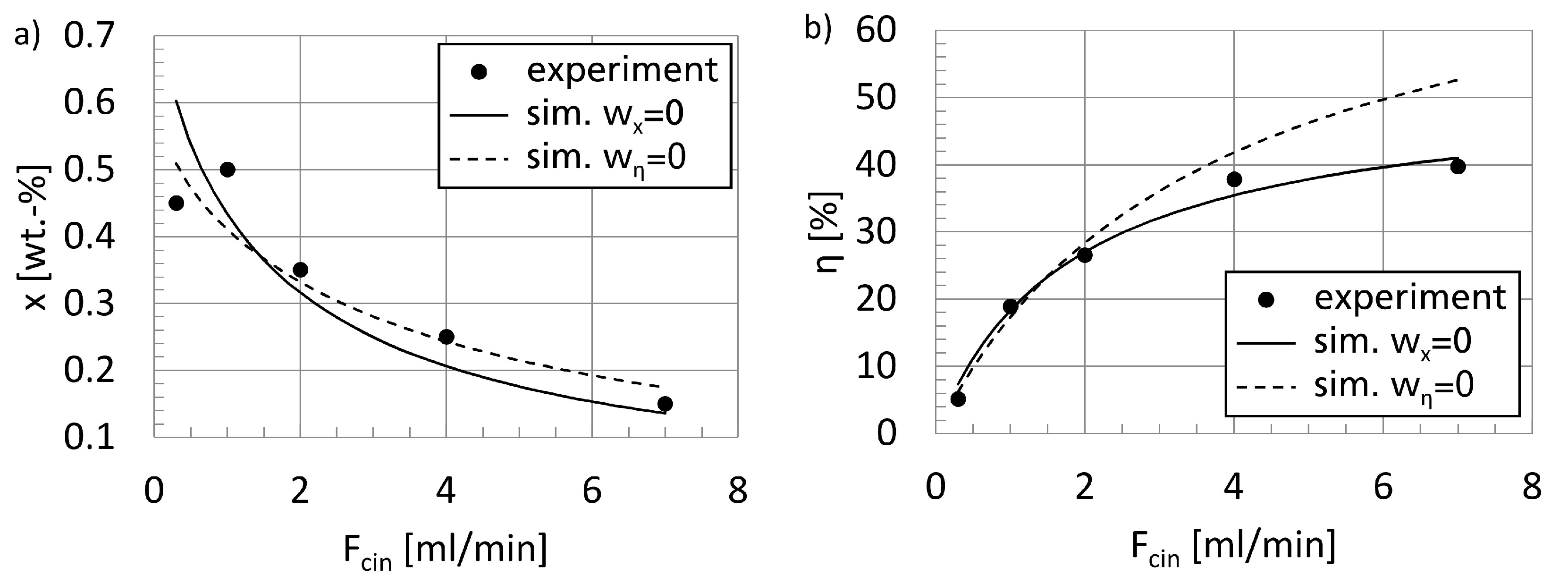
Figure 1. Alternatively, the kinetic parameters can be calculated by direct adjustment of the unit model to experimental data, as in this work.
The process model proposed in this article is set up in Aspen Plus V10
®. However, this commercial flowsheet simulator does not provide a model of an electrochemical reactor as a unit operation and hence, an electrolysis cell model was implemented in Aspen custom modeler V10
® (ACM) here. Note that this has been done before by Redissi and Bouallou [
35], who performed an economic evaluation of a high-temperature co-electrolysis process for conversion of CO
/H
O to synthesis gas, based on simulations in Aspen Plus
®. Apart from that, the process comprises a membrane distillation module [
36] for separation of acidic components from the product solution in the downstream processing, which is neither available in Aspen Plus
®. Implementations of such unit operations in Aspen Plus
® can be found in [
37,
38]. A critical review on the modeling of membrane distillation technologies was given by Hitsov et al. [
39]. A species-selective flow through a membrane is forced by the thermal gradient between a heated feed side and a cooler permeate side. The pores are hydrophobic and therefore the liquid phases from each side only partially penetrate the membrane. Material transport through the gas-filled segments of the pores is initiated by the difference in vapor pressures resulting from the different temperatures on both sides.
In most practical applications, electrochemical modules are part of higher-level processes with potential up- and downstream processing. However, the availability of studies considering electrochemistry on a process scale, i.e., in a coupled network of interacting modules rather than on the level of a unit operation, is limited. One of the few works on this topic was published by Karst et al. [
40], who employed a multi-scale approach to design a fuel cell process with methane reforming in the upstream part for automotive application. Furthermore, Sanchéz et al. modeled an electrolysis process for hydrogen production in Aspen Plus
® [
41]. The current article contributes to this point by applying multi-criteria optimization (MCO) [
42,
43] with an adaptive scalarization scheme to a process containing an electrochemical reactor. The resulting optimal operating points apply for the entire process rather than a single device. For the MCO, Aspen Plus V10
® is controlled via a programming interface. For this purpose, it offers an ActiveX automation server to be used within a VBA (visual basic for applications) code or a VB (visual basic) code. Previous work exploiting this interface for the optimization of separation processes can be found in [
44,
45]. The VBA code from [
42] is used in the current work, since it already comes with an automated graphical user interface in MS Excel. Only the flowsheet simulator is exchanged here.
The model equations for the electrochemical reactor and the membrane distillation module are discussed in detail in
Section 2.1 and
Section 2.2 of this article. Subsequently, in
Section 3.1, particular attention is paid to the determination of electro-kinetic rate constants for the reactor model. This is realized here by direct adjustment of the model to experimental data, using MS Excel as a central interface coupling the simulation with an external optimization solver. Since two measured variables are available, the adjustment procedure is treated as an MCO problem, as suggested in [
46]. A validation of the parametrized model equations is presented, too. Finally, according to the second modeling step in
Figure 1, the adjusted model is embedded in a flowsheet for the entire process in
Section 3.2. Pareto-optimal working points are then adaptively calculated [
43,
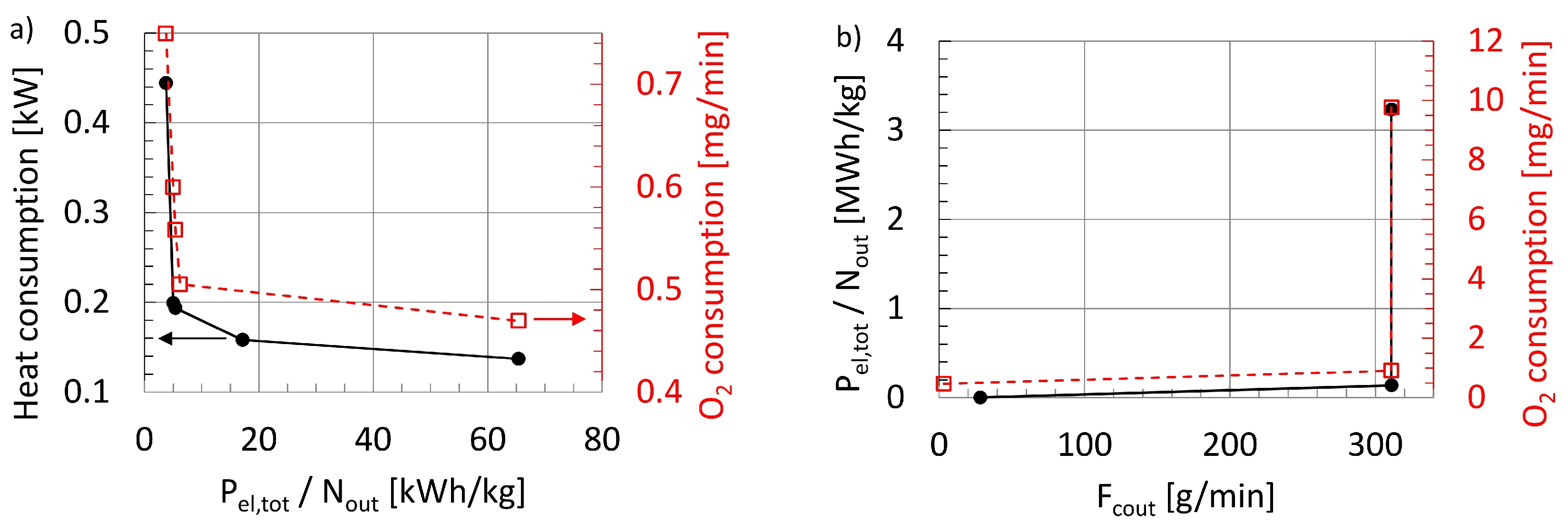
47], with particular respect to the electrical power required per product amount and the heat input.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}