
Histologic Patterns and Clues to Autoinflammatory Diseases in Children: What a Cutaneous Biopsy Can Tell Us


Abstract
:1. Introduction
1.1. Innate versus Adaptive Immunity: Autoinflammatory Versus Autoimmune Disease—What the Dermatopathologist Needs to Know
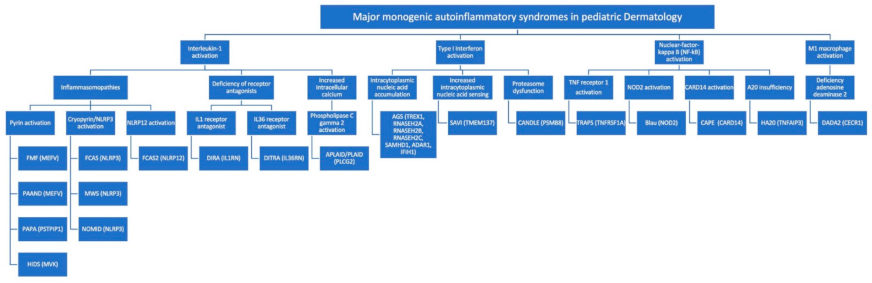
- IL-1 activation. The binding of an antigen to a pattern recognition receptor activates a pro-inflammatory cascade of intracellular, multimeric protein complexes called ‘inflammasomes.’ Inflammasomes are defined by their sensor proteins, which oligomerize to activate caspase-1, also called IL-1–converting enzyme, leading to proteolytic activation of IL-1b. Multiple inflammasomes are well-understood, including pyrin and cryopyrin. Cryopyrin is also called NLR family pyrin domain containing 3 (NLRP3). MAIS resulting from mutations within inflammasomes are also called ‘inflammasomopathies’. Unleashed IL1-induced inflammation can also result from deficiency of IL1 and IL36 receptor antagonists. Phospholipase C gamma 2 (PLCƔ2) is a cytoplasmic signaling enzyme, which, when recruited to the membrane upon receptor activation, induces the release of endoplasmic reticulum calcium stores, thereby leading to increased intracellular calcium levels and activation of the NLRP3 inflammasome.
- Type I interferon (IFN) activation (Type I interferonopathies). Autoinflammatory diseases related to IFN activation, also called interferonopathies, reflect aberrant activation of type I IFN pathways (IFN-α and IFN-β), which are involved in antiviral defense. Type I IFN production is triggered by viral RNA or DNA, and interferonopathies may arise through disorders of intracytoplasmic accumulation of endogenous nucleic acid due to their decreased degradation, through inherent, increased intracytoplasmic nucleic acid sensing or through a proteasome dysfunction.
- NF-κB activation (NFkBopathies). The NF-κB complex is a central signaling hub within the cytoplasm, integrating signals from multiple cell-surface receptors, including TNF receptors and intracellular pattern recognition receptors, like the nucleotide-binding oligomerization domain 2 (NOD2) receptor. NF-κB allows the freeing of several transcription factors, which move to the nucleus and trigger expression of proinflammatory genes. Activation of caspase-activating recruitment domain, member 14 (CARD14) also leads to enhanced NF-κB activity. A20 is a negative regulator of NF-κB and A20 insufficiency also results in an NFkBopathy.
- M1 macrophage activation. Adenosine deaminase 2 deficiency results in increased pro-inflammatory M1 macrophages (as opposed to anti-inflammatory M2 macrophages).
1.2. Histopathological Clues to the Diagnosis of Autoinflammation
2. Autoinflammatory Diseases: Correlating Histologic Patterns with Specific Diseases
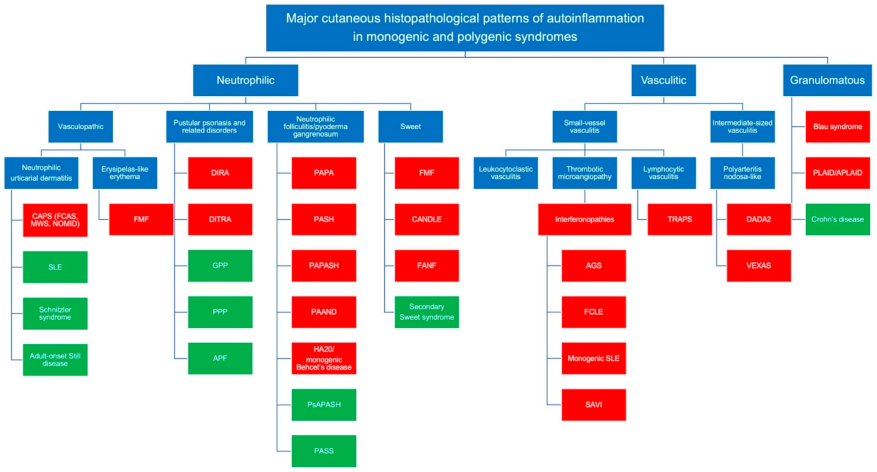
2.1. The Neutrophilic Pattern
2.1.1. The Vasculopathic Pattern
2.1.2. The Pustular Psoriasis Pattern
2.1.3. The Aseptic Neutrophilic Folliculitis Pattern
2.1.4. Sweet’s Syndrome
2.2. The Vasculitic Pattern
2.2.1. Small Sized-Vessel Vasculitis
2.2.2. Intermediate-Sized Vessel Vasculitis
2.3. The Granulomatous Pattern
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APLAID | autoinflammation and phospholipase C gamma 2 associated antibody deficiency and immune dysregulation |
| CANDLE | Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature |
| CAPE | caspase-activating recruitment domain, member 14 associated papulosquamous eruption |
| CAPS | cryopyrin-associated periodic syndromes |
| DADA2 | deficiency of adenosine deaminase 2 |
| DIRA | Deficiency of IL-1 Receptor Antagonist syndrome |
| DITRA | Deficiency of IL-36 Receptor Antagonist |
| FCAS | Familial Cold Autoinflammatory syndrome |
| FMF | Familial Mediterranean Fever syndrome |
| HA20 | Haploinsufficiency of A20 |
| HIDS | Hyperimmunoglobulinemia D syndrome |
| IFN | interferon |
| IL | interleukin |
| LE | lupus erythematosus |
| MAIS | monogenic autoinflammatory syndromes |
| MWS | Muckle–Wells syndrome |
| NOMID | neonatal-onset multisystem inflammatory disease |
| PAAND | pyrin-associated autoinflammation with neutrophilic dermatosis |
| PAPA | Pyogenic Arthritis, Pyoderma gangrenosum and Acne |
| PAPASH | Pyogenic Arthritis, Pyoderma gangrenosum, Acne, Suppurative Hidradenitis |
| PASH | Pyoderma gangrenosum, Acne, Suppurative Hidradenitis |
| PASS | Pyoderma gangrenosum, Acne, Suppurative hidradenitis and ankylosing Spondylitis |
| PLAID | Phospholipase C gamma 2 associated antibody deficiency and immune dysregulation |
| PLCƔ2 | Phospholipase C gamma 2 |
| PsAPASH | Psoriatic Arthritis, Pyoderma gangrenosum, Acne, Suppurative Hidradenitis |
| SAVI | Stimulator of interferon genes (STING) associated vasculopathy of infancy |
| TRAPS | TNF receptor-associated periodic syndrome |
| VEXAS | Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic syndrome |
References
- Nigrovic, P.A.; Lee, P.Y.; Hoffman, H.M. Monogenic autoinflammatory disorders: Conceptual overview, phenotype, and clinical approach. J. Allergy Clin. Immun. 2020, 146, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, S.; Kastner, D.L. Familial autoinflammatory diseases: Genetics, pathogenesis and treatment. Curr. Opin. Rheumatol. 2005, 17, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Chitkara, P.; Stojanov, S.; Kastner, D.L. The hereditary autoinflammatory syndromes. Pediatr. Infect. Dis. J. 2007, 26, 353–354. [Google Scholar] [CrossRef]
- McDermott, M.F.; Aksentijevich, I. The autoinflammatory syndromes. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Figueras-Nart, I.; Mascaró, J.M.; Solanich, X.; Hernández-Rodríguez, J. Dermatologic and Dermatopathologic Features of Monogenic Autoinflammatory Diseases. Front. Immunol. 2019, 10, 2448. [Google Scholar] [CrossRef]
- Shwin, K.W.; Lee, C.R.; Goldbach-Mansky, R. Dermatologic Manifestations of Monogenic Autoinflammatory Diseases. Derm. Clin. 2017, 35, 21–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipsker, D.; Saurat, J.-H. Neutrophilic cutaneous lupus erythematosus. At the edge between innate and acquired immunity? Dermatology 2008, 216, 283–286. [Google Scholar] [CrossRef]
- Stankovic, K.; Grateau, G. What’s new in autoinflammatory diseases? Rev. Med. Interne 2008, 29, 994–999. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Cowen, E.W.; Leslie, K.S. Autoinflammation: From monogenic syndromes to common skin diseases. J. Am. Dermatol. 2013, 68, 1–20. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekaert, S.M.C.; Böer-Auer, A.; Kerl, K.; Herrgott, I.; Schulz, X.; Bonsmann, G.; Brehler, R.; Metze, D. Neutrophilic Epitheliotropism Is a Histopathological Clue to Neutrophilic Urticarial Dermatosis. Am. J. Dermatopathol. 2016, 38, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Kolivras, A.; Theunis, A.; Ferster, A.; Lipsker, D.; Sass, U.; Dussart, A.; André, J. Cryopyrin-associated periodic syndrome: An autoinflammatory disease manifested as neutrophilic urticarial dermatosis with additional perieccrine involvement. J. Cutan. Pathol. 2011, 38, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Gusdorf, L.; Lipsker, D. Neutrophilic urticarial dermatosis. An entity bridging monogenic and polygenic autoinflammatory disorders, and beyond. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Gusdorf, L.; Lipsker, D. Neutrophilic urticarial dermatosis: A review. Ann. Dermatol. Vénéréologie 2018, 145, 735–740. [Google Scholar] [CrossRef]
- Aksentijevich, I.; Putnam, C.D.; Remmers, E.F.; Mueller, J.L.; Le, J.; Kolodner, R.D. The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheumatism. 2007, 56, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Neven, B.; Prieur, A.-M.; Maire, P.Q.D. Cryopyrinopathies: Update on pathogenesis and treatment. Nat. Clin. Pract. Rheumatol. 2008, 4, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Wanderer, A.A.; Broide, D.H. Familial cold autoinflammatory syndrome: Phenotype and genotype of an autosomal dominant periodic fever. J. Allergy Clin. Immunol. 2001, 108, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Jeru, I.; Duquesnoy, P.; Fernandes-Alnemri, T.; Cochet, E.; Yu, J.-W.; Lackmy-Port-Lis, M.; Grimprel, E.; Landman-Parker, J.; Hentgen, V.; Marlin, S.; et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA 2008, 105, 1614–1619. [Google Scholar] [CrossRef] [Green Version]
- Kilcline, C.; Shinkai, K.; Bree, A.; Modica, R.; Scheven, E.V.; Frieden, I.J. Neonatal-Onset multisystem inflammatory disorder: The emerging role of pyrin genes in autoinflammatory diseases. Arch. Dermatol. 2005, 141, 248–253. [Google Scholar] [CrossRef] [Green Version]
- Goldbach-Mansky, R.; Dailey, N.J.; Canna, S.W.; Gelabert, A.; Jones, J.; Rubin, B.I.; Kastner, D.L. Neonatal-Onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 2006, 355, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Shinkai, K.; McCalmont, T.H.; Leslie, K.S. Cryopyrin-associated periodic syndromes and autoinflammation. Clin. Exp. Dermatol. 2008, 33, 1–9. [Google Scholar] [CrossRef]
- Brinster, N.K.; Nunley, J.; Pariser, R.; Horvath, B. Nonbullous neutrophilic lupus erythematosus: A newly recognized variant of cutaneous lupus erythematosus. J. Am. Acad. Dermatol. 2012, 66, 92–97. [Google Scholar] [CrossRef]
- Sokumbi, O.; Drage, L.A.; Peters, M.S. Clinical and histopathologic review of Schnitzler syndrome: The Mayo Clinic experience (1972–2011). J. Am. Dermatol. 2012, 67, 1289–1295. [Google Scholar] [CrossRef]
- Santa, E.; McFalls, J.M.; Sahu, J.; Lee, J.B. Clinical and histopathological features of cutaneous manifestations of adult-onset Still disease. J. Cutan. Pathol. 2017, 44, 591–595. [Google Scholar] [CrossRef]
- Livneh, A.; Langevitz, P.; Zemer, D.; Zaks, N.; Kees, S.; Lidar, T.; Migdal, A.; Padeh, S.; Pras, M. Criteria for the diagnosis of familial mediterranean fever. Arthritis Rheum. 1997, 40, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Tamir, N.; Langevitz, P.; Zemer, D.; Pras, E.; Shinar, Y.; Padeh, S. Late-Onset familial Mediterranean fever (FMF): A subset with distinct clinical, demographic, and molecular genetic characteristics. Am. J. Med. Genet. 1999, 87, 30–35. [Google Scholar] [CrossRef]
- Padeh, S.; Livneh, A.; Pras, E.; Shinar, Y.; Lidar, M.; Feld, O.; Berkun, Y. Familial Mediterranean Fever in the First Two Years of Life: A Unique Phenotype of Disease in Evolution. J. Pediatr. 2010, 156, 985–989. [Google Scholar] [CrossRef]
- Lidar, M.; Doron, A.; Barzilai, A.; Feld, O.; Zaks, N.; Livneh, A.; Langevitz, P. Erysipelas-like erythema as the presenting feature of familial Mediterranean fever. J. Eur. Acad. Dermatol. Venereol. 2012, 27, 912–915. [Google Scholar] [CrossRef]
- Kolivras, A.; Provost, P.; Thompson, C.T. Erysipelas-like erythema of familial Mediterranean fever syndrome: A case report with emphasis on histopathologic diagnostic clues. J. Cutan. Pathol. 2013, 40, 585–590. [Google Scholar] [CrossRef]
- Barzilai, A.; Langevitz, P.; Goldberg, I.; Kopolovic, J.; Livneh, A.; Pras, M.; Trau, H. Erysipelas-like erythema of familial Mediterranean fever: Clinicopathologic correlation. J. Am. Acad. Dermatol. 2000, 42, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Akman, A.; Cakcak, D.S.; Çoban, E.; Ozbudak, H.I.; Ciftcioglu, M.A.; Alpsoy, E.; Yilmaz, E. Recurrent bullous lesions associated with familial Mediterranean fever: A case report. Clin. Exp. Dermatol. 2009, 34, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Aksentijevich, I.; Masters, S.L.; Ferguson, P.J.; Dancey, P.; Frenkel, J.; Van Royen-Kerkhoff, A.; Laxer, R.; Tedgård, U.; Cowen, E.W.; Pham, T.-H.; et al. An Autoinflammatory Disease with Deficiency of the Interleukin-1–Receptor Antagonist. N. Engl. J. Med. 2009, 360, 2426–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minkis, K.; Aksentijevich, I.; Goldbach-Mansky, R.; Magro, C.; Scott, R.; Davis, J.G.; Sardana, N.; Herzog, R. Interleukin 1 Receptor Antagonist Deficiency Presenting as Infantile Pustulosis Mimicking Infantile Pustular Psoriasis. Arch. Dermatol. 2012, 148, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.-Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36–Receptor Antagonist Deficiency and Generalized Pustular Psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Bachelez, H. Pustular psoriasis and related pustular skin diseases. Br. J. Dermatol. 2018, 178, 614–618. [Google Scholar] [CrossRef]
- Spoerri, I.; Herms, S.; Eytan, O.; Sarig, O.; Heinimann, K.; Sprecher, E.; Itin, P.; Burger, B. Immune-regulatory genes as possible modifiers of familial pityriasis rubra pilaris—Lessons from a family with PRP and psoriasis. J. Eur. Acad. Dermatol. Venereol. 2018, 32, e389–e392. [Google Scholar] [CrossRef]
- Sugiura, K. Autoinflammatory diseases in dermatology: DITRA and CAMPS. Nihon Rinsho Men’eki Gakkai Kaishi Jpn. J. Clin. Immunol. 2017, 40, 169–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craiglow, B.G.; Boyden, L.M.; Hu, R.; Virtanen, M.; Su, J.; Rodriguez, G.; McCarthy, C.; Luna, P.; Larralde, M.; Humphrey, S.; et al. CARD14-associated papulosquamous eruption: A spectrum including features of psoriasis and pityriasis rubra pilaris. J. Am. Acad. Dermatol. 2018, 79, 487–494. [Google Scholar] [CrossRef]
- Takeichi, T.; Sugiura, K.; Nomura, T.; Sakamoto, T.; Ogawa, Y.; Oiso, N.; Akiyama, M. Pityriasis Rubra Pilaris Type V as an Autoinflammatory Disease by CARD14 Mutations. JAMA Dermatol. 2017, 153, 66–70. [Google Scholar] [CrossRef]
- Ring, N.G.; Craiglow, B.G.; Panse, G.; Antaya, R.J.; Ashack, K.; Ashack, R.; Faith, E.F.; Paller, A.S.; McNiff, J.M.; Choate, K.A.; et al. Histopathologic findings characteristic of CARD14-associated papulosquamous eruption. J. Cutan. Pathol. 2020, 47, 425–430. [Google Scholar] [CrossRef]
- Schissler, C.; Velter, C.; Lipsker, D. Amicrobial pustulosis of the folds: Where have we gone 25 years after its original description? Ann. Dermatol. Vénéréol. 2017, 144, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, J.; Madrange, M.; Gardair, C.; Sbidian, E.; Frazier, A.; Wolkenstein, P.; Bachelez, H. PAPASH, Ps APASHand PASSautoinflammatory syndromes: Phenotypic heterogeneity, common biological signature and response to immunosuppressive regimens. Br. J. Dermatol. 2019, 181, 866–869. [Google Scholar] [CrossRef]
- Zhang, X.; He, Y.; Xu, H.; Wang, B. First PSENEN mutation in PASH syndrome. J. Dermatol. 2020, 47, 1335–1337. [Google Scholar] [CrossRef]
- Marzano, A.V.; Trevisan, V.; Gattorno, M.; Ceccherini, I.; de Simone, C.; Crosti, C. Pyogenic Arthritis, Pyoderma Gangrenosum, Acne, and Hidradenitis Suppurativa (PAPASH): A New Autoinflammatory Syndrome Associated with a Novel Mutation of the PSTPIP1 Gene. JAMA Dermatol. 2013, 149, 762–764. [Google Scholar] [CrossRef]
- Tallon, B.; Corkill, M. Peculiarities of PAPA syndrome. Rheumatology 2006, 45, 1140–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.J.; Allantaz, F.; Bennett, L.; Zhang, N.; Gao, X.; Wood, G.; Kastner, D.L.; Punaro, M.; Aksentijevich, I.; Pascual, V.; et al. Clinical, Molecular, and Genetic Characteristics of PAPA Syndrome: A Review. Curr. Genom. 2010, 11, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasini, C.; Michelerio, A. Erosive pustular dermatosis of the scalp: A neutrophilic folliculitis within the spectrum of neutrophilic dermatoses: A clinicopathologic study of 30 cases. J. Am. Acad. Dermatol. 2018, 81, 527–533. [Google Scholar] [CrossRef]
- Masters, S.L.; Lagou, V.; Jéru, I.; Baker, P.J.; Van Eyck, L.; Parry, D.A.; Lawless, D.; De Nardo, D.; Garcia-Perez, J.E.; Dagley, L.F.; et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl. Med. 2016, 8, 332ra45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aeschlimann, F.A.; Batu, E.D.; Canna, S.W.; Go, E.; Gül, A.; Hoffmann, P.; Laxer, R.M. A20 haploinsufficiency (HA20): Clinical phenotypes and disease course of patients with a newly recognised NF-κB-mediated autoinflammatory disease. Ann. Rheum. Dis. 2018, 77, 728. [Google Scholar] [CrossRef] [PubMed]
- Kaustio, M.; Haapaniemi, E.; Göös, H.; Hautala, T.; Park, G.; Syrjänen, J.; Einarsdottir, E.; Sahu, B.; Kilpinen, S.; Rounioja, S.; et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J. Allergy Clin. Immunol. 2017, 140, 782–796. [Google Scholar] [CrossRef] [Green Version]
- Oskay, T.; Anadolu, R. Sweet’s syndrome in familial Mediterranean fever: Possible continuum of the neutrophilic reaction as a new cutaneous feature of FMF. J. Cutan. Pathol. 2009, 36, 901–905. [Google Scholar] [CrossRef]
- Torrelo, A.; Patel, S.; Colmenero, I.; Gurbindo, D.; Lendinez, F.; Hernandez, A.; Paller, A.S. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J. Am. Acad. Dermatol. 2010, 62, 489–495. [Google Scholar] [CrossRef]
- Sanchez, I.M.; Lowenstein, S.; Johnson, K.A.; Babik, J.; Haag, C.; Keller, J.J.; Shinkai, K. Clinical Features of Neutrophilic Dermatosis Variants Resembling Necrotizing Fasciitis. JAMA Dermatol. 2019, 155, 79–82. [Google Scholar] [CrossRef]
- Kaustio, M.; Hautala, T.; Seppänen, M.R.J. Primary Immunodeficiency, a Possible Cause of Neutrophilic Necrotizing Dermatosis. JAMA Dermatol. 2019, 155, 863–864. [Google Scholar] [CrossRef]
- Jain, A.; Misra, D.P.; Sharma, A.; Wakhlu, A.; Agarwal, V.; Negi, V.S. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol. Int. 2018, 38, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Durel, C.-A.; Aouba, A.; Bienvenu, B.; Deshayes, S.; Coppéré, B.; Gombert, B.; Hot, A. Observational Study of a French and Belgian Multicenter Cohort of 23 Patients Diagnosed in Adulthood with Mevalonate Kinase Deficiency. Medicine 2016, 95, e3027. [Google Scholar] [CrossRef]
- Korppi, M.; Van Gijn, M.E.; Antila, K. Hyperimmunoglobulinemia D and periodic fever syndrome in children. Review on therapy with biological drugs and case report. Acta Paediatr. 2011, 100, 21–25. [Google Scholar] [CrossRef]
- Drenth, J.P.; Boom, B.W.; Toonstra, J.; van der Meer, J.W. Cutaneous manifestations and histologic findings in the hyperimmunoglobulinemia D syndrome. International Hyper IgD Study Group. Arch. Dermatol. 1994, 130, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, S.; Kitamura, W.; Morita, K.; Saishin, M.; Shirai, T. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br. J. Dermatol. 1993, 128, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Boom, B.W.; Daha, M.R.; Vermeer, B.J.; van der Meer, J.W. IgD immune complex vasculitis in a patient with hyperimmunoglobulinemia D and periodic fever. Arch. Dermatol. 1990, 126, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Munoz, J.; Marque, M.; Dandurand, M.; Meunier, L.; Crow, Y.J.; Bessis, D. Interféronopathies de type I. Ann. Dermatol. Vénéréol. 2015, 142, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Omarjee, O.; Picard, C.; Frachette, C.; Moreews, M.; Laucat, F.R.; Sprauel, P.; Belot, A. Monogenic lupus: Dissecting heterobeneity. Autoimmun. Rev. 2019, 18, 102361. [Google Scholar] [CrossRef] [PubMed]
- Günther, C.; Berndt, N.; Wolf, C.; Lee-Kirsch, M.A. Familial Chilblain Lupus Due to a Novel Mutation in the Exonuclease III Domain of 3′ Repair Exonuclease 1 (TREX1). JAMA Dermatol. 2015, 151, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Beltoise, A.S.; Audouin-Pajot, C.; Lucas, P.; Tournier, E.; Rice, G.I.; Crow, Y.J.; Mazereeuw-Hautier, J. Lupus-engelures familial: Quatre cas sur trois générations. Ann. Dermatol. Vénéréol. 2018, 145, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Kolivras, A.; Aeby, A.; Crow, Y.J.; Rice, G.I.; Sass, U.; André, J. Cutaneous histopathological findings of Aicardi-Goutières syndrome, overlap with chilblain lupus. J. Cutan. Pathol. 2008, 35, 774–778. [Google Scholar] [CrossRef]
- Lee-Kirsch, M.A.; Wolf, C.; Günther, C. Aicardi-Goutières syndrome: A model disease for systemic autoimmunity. Clin. Exp. Immunol. 2014, 175, 17–24. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Montealegre Sanchez, G.A.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a Vascular and Pulmonary Syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Munoz, J.; Rodière, M.; Jeremiah, N.; Rieux-Laucat, F.; Oojageer, A.; Rice, G.I.; Rozenberg, F.; Crow, Y.J.; Bessis, D. Stimulator of Interferon Genes–Associated Vasculopathy With Onset in Infancy. JAMA Dermatol. 2015, 151, 872–877. [Google Scholar] [CrossRef]
- Crow, Y.J.; Casanova, J.-L. STING-Associated Vasculopathy with Onset in Infancy—A New Interferonopathy. N. Engl. J. Med. 2014, 371, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.; Eroglu, F.K.; Özen, S.; Orhan, D.; Montealegre-Sanchez, G.; De Jesus, A.A.; Goldbach-Mansky, R.; Cowen, E.W. Failure to thrive, interstitial lung disease, and progressive digital necrosis with onset in infancy. J. Am. Acad. Dermatol. 2016, 74, 186–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojanov, S.; McDermott, M.F. the tumour necrosis factor receptor-associated periodic syndrome: Current concepts. Expert Rev. Mol. Med. 2005, 7, 1–18. [Google Scholar] [CrossRef]
- Masson, C.; Simon, V.; Hoppe, E.; Insalaco, P.; Cissé, I.; Audran, M. Tumor necrosis factor receptor-associated periodic syndrome (TRAPS): Definition, semiology, prognosis, pathogenesis, treatment, and place relative to other periodic joint diseases. Jt. Bone Spine 2004, 71, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Toro, J.R.; Aksentijevich, I.; Hull, K.; Dean, J.; Kastner, D.L. Tumor necrosis factor receptor-associated periodic syndrome: A novel syndrome with cutaneous manifestations. Arch. Dermatol. 2000, 136, 1487–1494. [Google Scholar] [CrossRef] [Green Version]
- Farasat, S.; Aksentijevich, I.; Toro, J.R. Autoinflammatory Diseases: Clinical and Genetic Advances. Arch. Dermatol. 2008, 144, 392–402. [Google Scholar] [CrossRef]
- Elkan, P.N.; Pierce, S.B.; Segel, R.; Walsh, T.; Barash, J.; Padeh, S.; Zlotogorski, A.; Berkun, Y.; Press, J.J.; Mukamel, M.; et al. Mutant Adenosine Deaminase 2 in a Polyarteritis Nodosa Vasculopathy. N. Engl. J. Med. 2014, 370, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Fayand, A.; Sarrabay, G.; Belot, A.; Hentgen, V.; Kone-Paut, I.; Grateau, G.; Georgin-Lavialle, S. Les multiples facettes du déficit en ADA2, vascularite, maladie auto-inflammatoire et immunodéficit: Mise au point à partir des 135 cas de la littérature. Rev. Méd. Interne 2018, 39, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, D.; Ombrello, A.K.; Zavialov, A.; Toro, C.; Zavialov, A.V.; Stone, D.L.; Chae, J.J.; Rosenzweig, S.D.; Bishop, K.; et al. Early-Onset Stroke and Vasculopathy Associated with Mutations in ADA2. N. Engl. J. Med. 2014, 370, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Caorsi, R.; Penco, F.; Grossi, A.; Insalaco, A.; Omenetti, A.; Alessio, M.; Conti, G.; Marchetti, F.; Picco, P.; Tommasini, A.; et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: A multicentre national study. Ann. Rheum. Dis. 2017, 76, 1648–1656. [Google Scholar] [CrossRef]
- Pichard, D.C.; Ombrello, A.K.; Hoffmann, P.; Stone, D.L.; Cowen, E.W. Early-onset stroke, polyarteritis nodosa (PAN), and livedo racemosa. J. Am. Acad. Dermatol. 2016, 75, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Santiago, T.M.G.; Zavialov, A.; Saarela, J.; Seppanen, M.; Reed, A.M.; Abraham, R.S.; Gibson, L.E. Dermatologic Features of ADA2 Deficiency in Cutaneous Polyarteritis Nodosa. JAMA Dermatol. 2015, 151, 1230–1234. [Google Scholar] [CrossRef] [Green Version]
- Caorsi, R.; Penco, F.; Schena, F.; Gattorno, M. Monogenic polyarteritis: The lesson of ADA2 deficiency. Pediatr. Rheumatol. 2016, 14, 51. [Google Scholar] [CrossRef] [Green Version]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Cardona, D.O.; Wu, Z.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N. Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, A.M.; Ombrello, M.J. Using genes to triangulate the pathophysiology of granulomatous autoinflammatory disease: NOD2, PLCG2 and LACC1. Int. Immunol. 2018, 30, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Li, E.; Shen, B. Autoinflammatory disease with focus on NOD2-associated disease in the era of genomic medicine. Autoimmunity 2019, 52, 48–56. [Google Scholar] [CrossRef]
- Wlodek, C.; Clinch, J.; Planas, S.; Shaw, L. Widespread papular eruption in an infant. Clin. Exp. Dermatol. 2018, 43, 212–215. [Google Scholar] [CrossRef]
- Caso, F.; Galozzi, P.; Costa, L.; Sfriso, P.; Cantarini, L.; Punzi, L. Autoinflammatory granulomatous diseases: From Blau syndrome and early-onset sarcoidosis to NOD2-mediated disease and Crohn’s disease. RMD Open 2015, 1, e000097. [Google Scholar] [CrossRef] [PubMed]
- Poline, J.; Fogel, O.; Pajot, C.; Miceli-Richard, C.; Rybojad, M.; Galeotti, C. Early onset granulomatous arthritis, uveitis and skin rash: Characterisation of skin involvement in Blau syndrome. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 340–348. [Google Scholar] [CrossRef]
- Janssen, C.E.I.; Rosé, C.D.; Hertogh, G.D.; Martin, T.M.; Bader-Meunier, B.; Cimaz, R.; Wouters, C.H. Morphologic and immunohistochemical characterization of granulomas in the nucleotide oligomerization domain 2-related disorders Blau syndrome and Crohn disease. J. Allergy Clin. Immunol. 2012, 129, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Su, L.-C.; Tomecki, K.J.; Zhou, L.; Jayakar, B.; Shen, B. Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J. Am. Acad. Dermatol. 2013, 68, 624–631. [Google Scholar] [CrossRef]
- Shen, M.; Moran, R.; Tomecki, K.J.; Yao, Q. Granulomatous disease associated with NOD2 sequence variants and familial camptodactyly: An intermediate form of NOD2-associated diseases? Semin. Arthritis Rheu. 2015, 45, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, M.; Marjańska, A.; Bąbol-Pokora, K.; Urbańczyk, A.; Grześk, E.; Młynarski, W.; Kołtan, S. Co-existence of Blau syndrome and NAID? Diagnostic challenges associated with presence of multiple pathogenic variants in NOD2 gene: A case report. Pediatr. Rheumatol. Online J. 2017, 15, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morán-Villaseñor, E.; Saez-De-Ocariz, M.; Torrelo, A.; Arostegui, J.I.; Yamazaki-Nakashimada, M.A.; Alcántara-Ortigoza, M.A.; González-Del-Angel, A.; Velázquez-Aragón, J.A.; López-Herrera, G.; Berrón-Ruiz, L.; et al. Expanding the clinical features of autoinflammation and phospholipase Cγ2-associated antibody deficiency and immune dysregulation by description of a novel patient. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 2334–2339. [Google Scholar] [CrossRef]
- Aderibigbe, O.M.; Priel, D.L.; Lee, C.-C.R.; Ombrello, M.J.; Prajapati, V.H.; Liang, M.G.; Milner, J.D. Distinct Cutaneous Manifestations and Cold-Induced Leukocyte Activation Associated with PLCG2 Mutations. JAMA Dermatol. 2015, 151, 627–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.D. PLAID: A Syndrome of Complex Patterns of Disease and Unique Phenotypes. J. Clin. Immunol. 2015, 35, 527–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ombrello, M.; Remmers, E.F.; Sun, G.; Freeman, A.F.; Datta, S.; Torabi-Parizi, P.; Subramanian, N.; Bunney, T.D.; Baxendale, R.W.; Martins, M.; et al. Cold Urticaria, Immunodeficiency, and Autoimmunity Related toPLCG2Deletions. N. Engl. J. Med. 2012, 366, 330–338. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
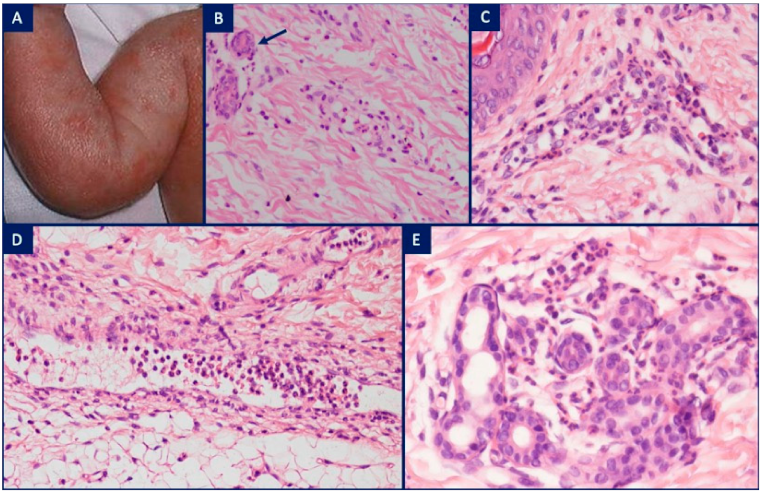
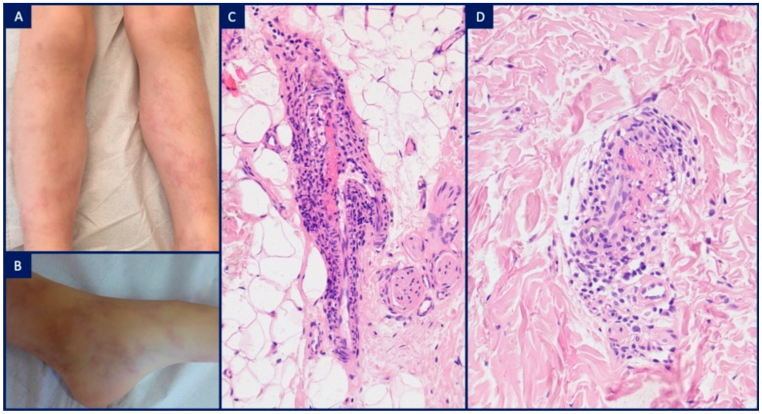
| Interleukin-1 activation | Inflammasomopathies | Pyrin activation | FMF (MEFV) | Erysipelas-like erythema in the only pathognomic cutaneous marker of FMF and is characterized by mild papillary dermal edema, dilated vessels, sparse perivascular mononuclear cell infiltrate admixed with neutrophils and nuclear dust |
| PAAND (MEFV) and PAPA (PSTPIP1) | Neutrophilic dermatosis comprising the phenotypical spectrum of pyoderma gangrenosum, neutrophilic folliculitis, abscess and acne, PAAND also shows small vessel leukocytoclastic vasculitis | |||
| Cryopyrin/NLRP3 activation | Cryopyrinopathies: FCAS, MWS, NOMID | Neutrophilic urticarial dermatitis: perivascular and interstitial neutrophilic infiltrate with variable leukocytoclasia, linear interstitial arrangement of neutrophils as ‘Indian file’, neutrophilic epitheliotropism including intraepidermal and peri-eccrine involvement, absence of fibrin in vessel walls, no significant dermal edema | ||
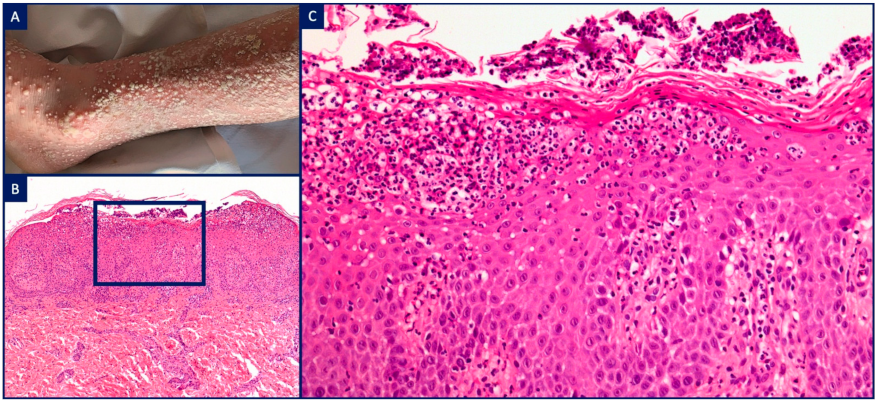
| IL1 receptor antagonist | DIRA (ILRN) | Pustular psoriasis (psoriasiform epidermal hyperplasia, subcorneal spongiform pustulation, neutrophilic aggregates within parakeratotic stratum corneum), dense intradermal neutrophilic infiltrate with peri-eccrine involvement | ||
| IL36 receptor antagonist | DITRA (IL36RN) | |||
| Phospholipase C gamma 2 activation | APLAID (PLCG2) | Dense perivascular and interstitial granulomatous infiltrate, palisading granulomas around necrobiotic collagen, dense neutrophilic infiltrate, intense papillary dermal oedema leading to subepidermal blistering | ||
| PLAID (PLCG2) | Neutrophilic urticarial dermatitis (cold-induced urticaria) and non-caseating sarcoidal granulomas | |||
| Type I Interferon activation | Intracytoplasmic nucleic acid accumulation | AGS (TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, IFIH1) and FCLE (TREX1, SAMHD1) | Thrombotic microangiopathy associated with findings of chilblain lupus erythematosus (vacuolar interface dermatitis, lymphocytic vasculitis, peri-eccrine lymphocytic infiltrate) | |
| Increased intracytoplasmic nucleic acid sensing | SAVI (TMEM137) | Thrombotic microangiopathy | ||
| Proteasome dysfunction | CANDLE (PSMB8) | Histiocytoid Sweet syndrome: dense perivascular and interstitial mononuclear cell infiltrate composed of immature myeloid cells admixed with mature neutrophils, eosinophils and leukocytoclasia | ||
| NF-κB activation | TNF receptor 1 activation | TRAPS (TNFRSF1A) | Lymphocytic vasculitis: perivascular lymphocytic infiltrate showing tight cuffing within both superficial and deep dermis, absence of fibrin within vessel walls | |
| NOD2 activation | Blau (NOD2) | Non-caseating sarcoidal granulomas with lymphocytic coronas | ||
| NAID (NOD2) | Subacute spongiotic dermatitis, irregular epidermal acanthosis with overlying parakeratotic hyperkeratosis | |||
| CARD14 activation | CAPE (CARD14) | Pityriasis rubra pilaris (psoriasiform epidermal hyperplasia, irregular hyperkeratosis, alternating vertical and horizontal orthokeratosis and parakeratosis, follicular plugging), absence of acantholysis | ||
| A20 insufficiency | HA20 (TNFAIP3) | Non-specific (oral aphtous erosions or ulcerations similarly to Behcet’s disease) | ||
| M1 macrophage activation | Deficiency adenosine deaminase 2 | DADA2 (CECR1) | Polyarteritis nodosa: neutrophilic infiltrate around and within muscular arteriole walls (intermediate-sized vessels at the dermo-hypodermal junction), disruption of the internal elastic lamina, fibrin deposition and intraluminal thrombosis | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolivras, A.; Meiers, I.; Sass, U.; Thompson, C.T. Histologic Patterns and Clues to Autoinflammatory Diseases in Children: What a Cutaneous Biopsy Can Tell Us. Dermatopathology 2021, 8, 202-220. https://doi.org/10.3390/dermatopathology8020026
Kolivras A, Meiers I, Sass U, Thompson CT. Histologic Patterns and Clues to Autoinflammatory Diseases in Children: What a Cutaneous Biopsy Can Tell Us. Dermatopathology. 2021; 8(2):202-220. https://doi.org/10.3390/dermatopathology8020026
Chicago/Turabian StyleKolivras, Athanassios, Isabelle Meiers, Ursula Sass, and Curtis T. Thompson. 2021. "Histologic Patterns and Clues to Autoinflammatory Diseases in Children: What a Cutaneous Biopsy Can Tell Us" Dermatopathology 8, no. 2: 202-220. https://doi.org/10.3390/dermatopathology8020026
APA StyleKolivras, A., Meiers, I., Sass, U., & Thompson, C. T. (2021). Histologic Patterns and Clues to Autoinflammatory Diseases in Children: What a Cutaneous Biopsy Can Tell Us. Dermatopathology, 8(2), 202-220. https://doi.org/10.3390/dermatopathology8020026