
Development of a New Method to Estimate the Water Purification Efficiency of Bulk-Supported Nanosorbents under Realistic Conditions



Abstract
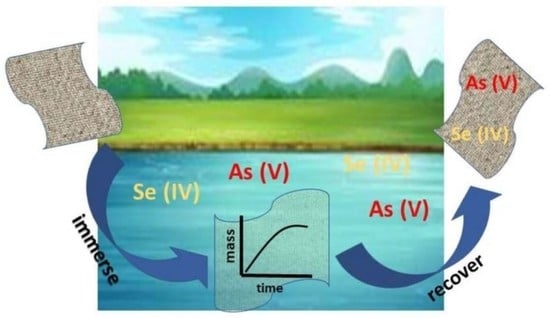
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Fabrication of MOR−1@Cotton Fabric
2.3. Characterization of the Sorbent
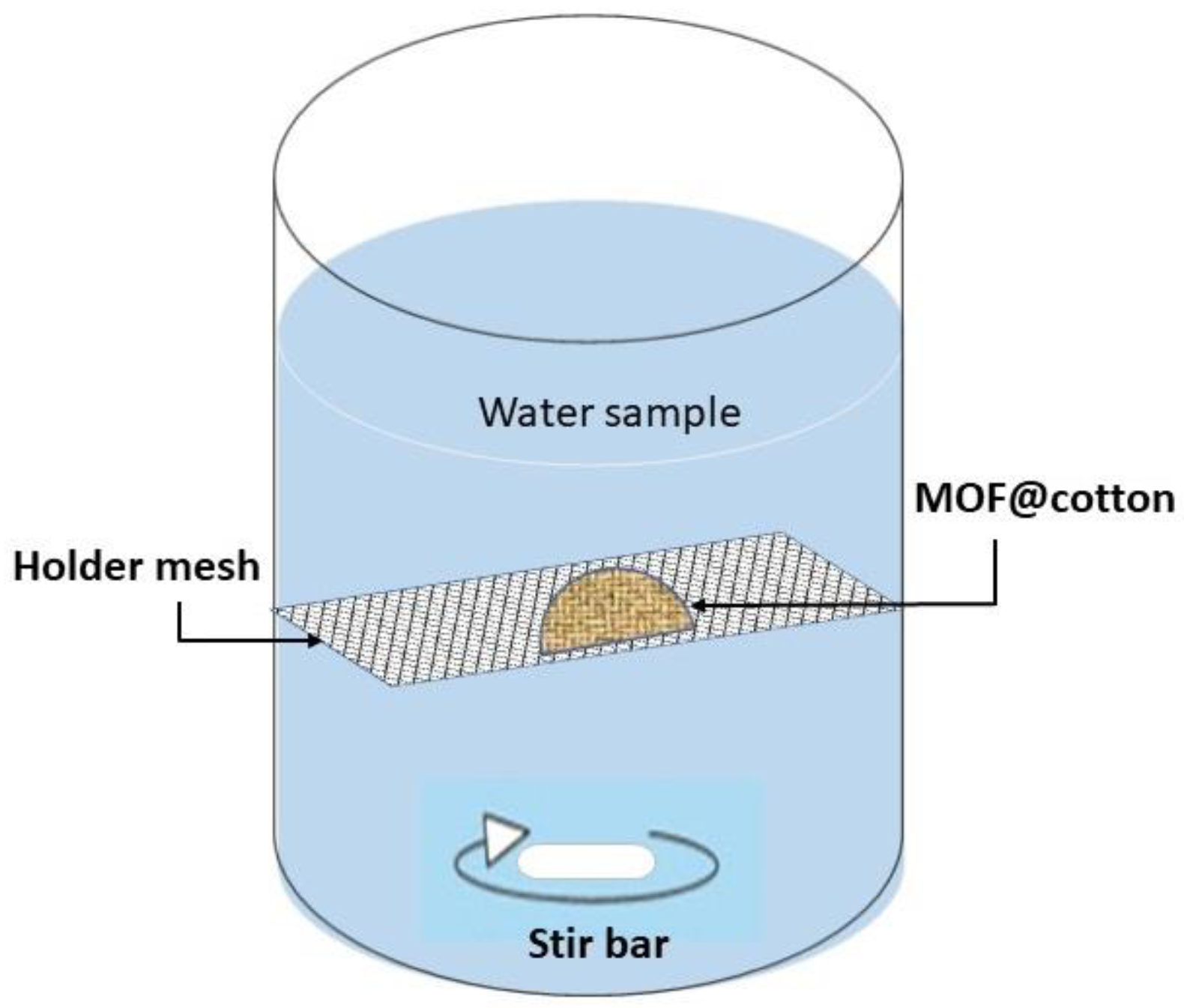
2.4. Experimental Procedure for Oxyanion Uptake Studies
3. Results
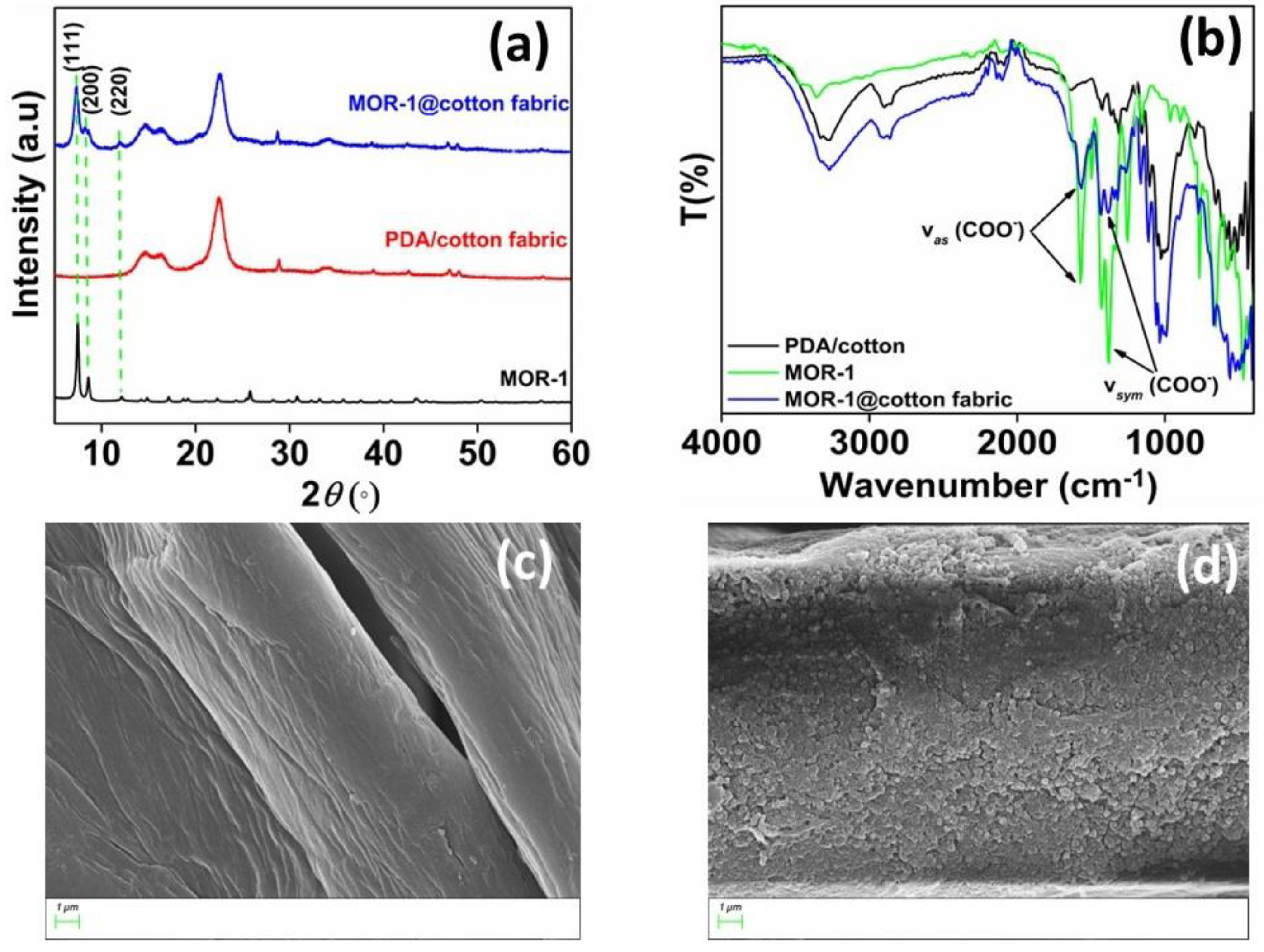
3.1. Characterization of MOR−1@Cotton Fabric
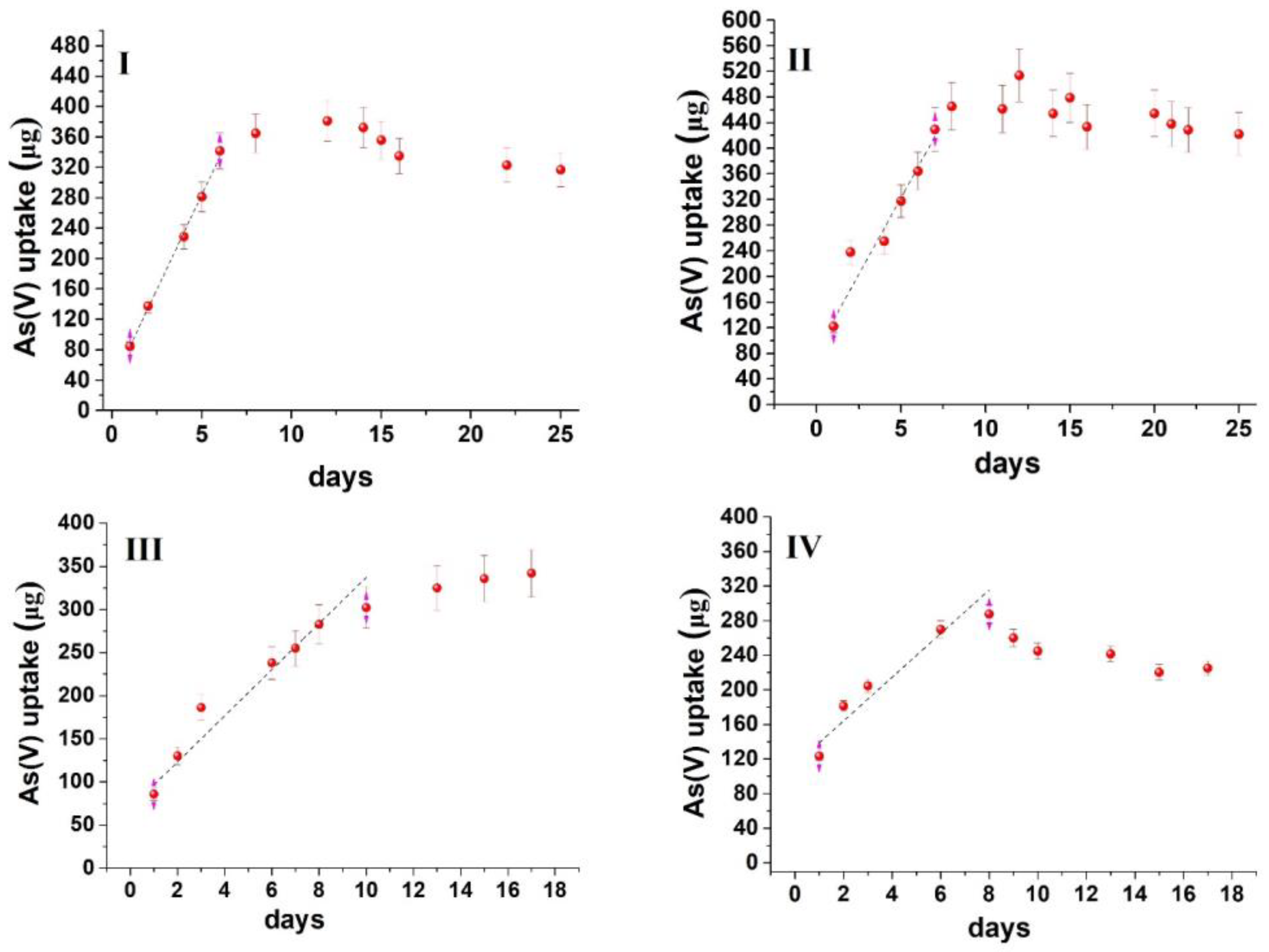
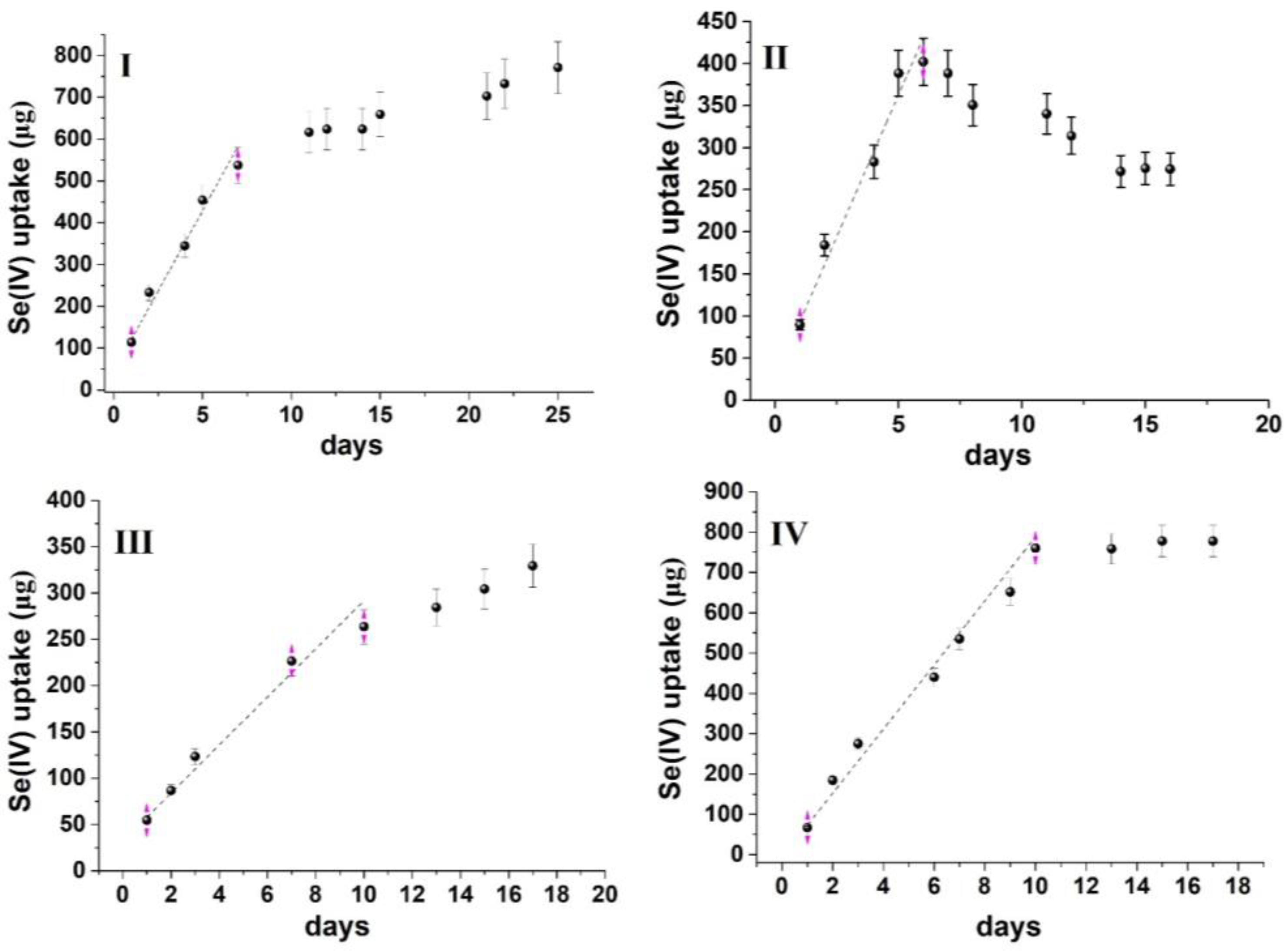
3.2. Principles of Passive Sampling for Modeling Contaminants’ Uptake by a Sorbent
3.3. Repurposing Passive Sampling (Diffusion) Modeling to Water Remediation
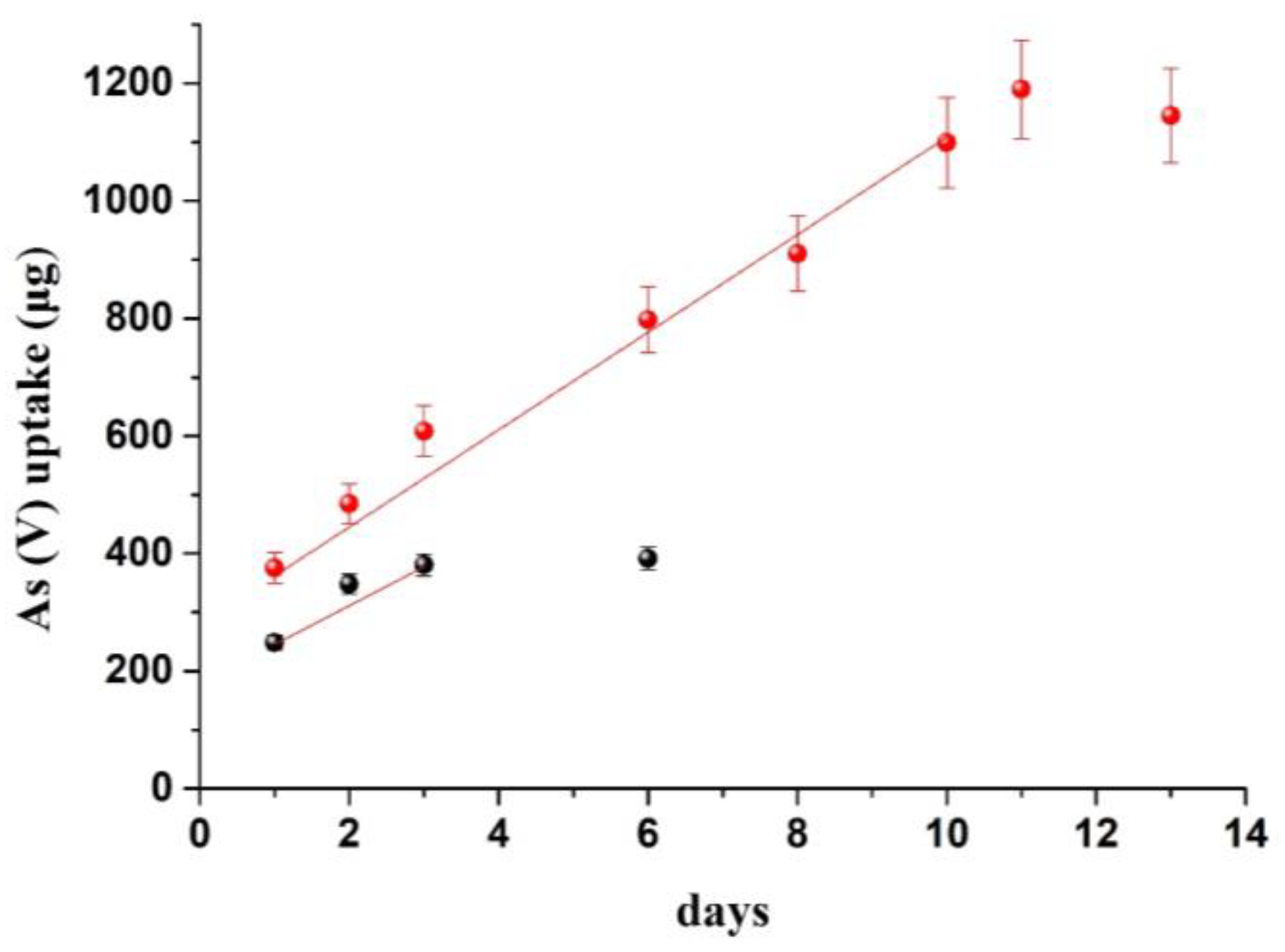
3.4. Method Demonstration
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grumezescu, A.M. (Ed.) Water Purification; Elsevier B.V.: Amsterdam, The Netherlands, 2017; ISBN 9780128043714. [Google Scholar]
- Lahnsteiner, J. (Ed.) Handbook of Water and Used Water Purification; Springer International Publishing: Cham, Switzerland, 2020; ISBN 978-3-319-66382-1. [Google Scholar]
- Singh, N.B.; Nagpal, G.; Agrawal, S.; Rachna. Water Purification by Using Adsorbents: A Review. Environ. Technol. Innov. 2018, 11, 187–240. [Google Scholar] [CrossRef]
- Santhosh, C.; Velmurugan, V.; Jacob, G.; Jeong, S.K.; Grace, A.N.; Bhatnagar, A. Role of Nanomaterials in Water Treatment Applications: A Review. Chem. Eng. J. 2016, 306, 1116–1137. [Google Scholar] [CrossRef]
- Lu, H.; Wang, J.; Stoller, M.; Wang, T.; Bao, Y.; Hao, H. An Overview of Nanomaterials for Water and Wastewater Treatment. Adv. Mater. Sci. Eng. 2016, 2016, 4964828. [Google Scholar] [CrossRef] [Green Version]
- Manos, M.J.; Kanatzidis, M.G. Metal Sulfide Ion Exchangers: Superior Sorbents for the Capture of Toxic and Nuclear Waste-Related Metal Ions. Chem. Sci. 2016, 7, 4804–4824. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Pournara, A.; Kim, K.-H.; Bansal, V.; Rapti, S.; Manos, M.J. Metal-Organic Frameworks: Challenges and Opportunities for Ion-Exchange/Sorption Applications. Prog. Mater. Sci. 2017, 86, 25–74. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhao, G.; Chen, C.; Chai, Z.; Alsaedi, A.; Hayat, T.; Wang, X. Metal–Organic Framework-Based Materials: Superior Adsorbents for the Capture of Toxic and Radioactive Metal Ions. Chem. Soc. Rev. 2018, 47, 2322–2356. [Google Scholar] [CrossRef]
- Gehrke, I.; Geiser, A.; Somborn-Schulz, A. Innovations in Nanotechnology for Water Treatment. Nanotechnol. Sci. Appl. 2015, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Westerhoff, P.; Alvarez, P.; Li, Q.; Gardea-Torresdey, J.; Zimmerman, J. Overcoming Implementation Barriers for Nanotechnology in Drinking Water Treatment. Environ. Sci. Nano 2016, 3, 1241–1253. [Google Scholar] [CrossRef]
- Mauter, M.S.; Zucker, I.; Perreault, F.; Werber, J.R.; Kim, J.-H.; Elimelech, M. The Role of Nanotechnology in Tackling Global Water Challenges. Nat. Sustain. 2018, 1, 166–175. [Google Scholar] [CrossRef]
- Borovik, A.; Karanikola, V.; Zucker, I. Platform Selection of Engineered Nanomaterials for Water Decontamination Applications. Environ. Sci. Nano 2020, 7, 3641–3654. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, H.; Yu, F.; Liu, Y.; Jia, L.; Zhao, W.; Li, P.; Wang, H.; Zhu, P.; Li, B. Blow Spinning of Polyimide/SiO2 Composite Fibrous Sponges with Excellent Adsorption Capacity and Recyclability. ACS Appl. Polym. Mater. 2022, 4, 8487–8495. [Google Scholar] [CrossRef]
- Bouraie, M.E.; Abdelghany, A. Sorption Features of Polyurethane Foam Functionalized with Salicylate for Chlorpyrifos: Equilibrium, Kinetic Models and Thermodynamic Studies. Polymers 2020, 12, 2036. [Google Scholar] [CrossRef]
- Pournara, A.D.; Moisiadis, E.; Gouma, V.; Manos, M.J.; Giokas, D.L. Cotton Fabric Decorated by a Zr4+ MOF for Selective As(V) and Se(IV) Removal from Aqueous Media. J. Environ. Chem. Eng. 2022, 10, 107705. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, G.; Dai, H.; Yang, M.; Fu, Y.; Ying, Y.; Li, Y. Biomineralization-Mimetic Preparation of Hybrid Membranes with Ultra-High Loading of Pristine Metal–Organic Frameworks Grown on Silk Nanofibers for Hazard Collection in Water. J. Mater. Chem. A 2018, 6, 3402–3413. [Google Scholar] [CrossRef]
- Godlewska, K.; Stepnowski, P.; Paszkiewicz, M. Pollutant Analysis Using Passive Samplers: Principles, Sorbents, Calibration and Applications. A Review. Environ. Chem. Lett. 2021, 19, 465–520. [Google Scholar] [CrossRef]
- Booij, K.; Vrana, B.; Huckins, J.N. Chapter 7. Theory, Modelling and Calibration of Passive Samplers Used in Water Monitoring. In Comprehensive Analytical Chemistry; Greenwood, R., Mills, G., Vrana, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 48, pp. 141–169. [Google Scholar]
- Gong, X.; Li, K.; Wu, C.; Wang, L.; Sun, H. Passive Sampling for Monitoring Polar Organic Pollutants in Water by Three Typical Samplers. Trends Environ. Anal. Chem. 2018, 17, 23–33. [Google Scholar] [CrossRef]
- Salim, F.; Górecki, T. Theory and Modelling Approaches to Passive Sampling. Environ. Sci. Process. Impacts 2019, 21, 1618–1641. [Google Scholar] [CrossRef]
- Gouma, V.; Pournara, A.D.; Manos, M.J.; Giokas, D.L. Fabric Phase Sorpitive Extraction and Passive Sampling of Ultraviolet Filters from Natural Waters Using a Zirconium Metal Organic Framework-Cotton Composite. J. Chromatogr. A 2022, 1670, 462945. [Google Scholar] [CrossRef]
- Gartner, J.W.; Wellman, R.E.; Wood, T.M.; Cheng, R.T. Water Velocity and Suspended Solids Measurements by In-Situ Instruments in Upper Klamath Lake, Oregon; U.S. Geological Survey: Reston, VA, USA, 2007. [Google Scholar]
- Schulze, K.; Hunger, M.; Döll, P. Simulating River Flow Velocity on Global Scale. Adv. Geosci. 2005, 5, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Rapti, S.; Pournara, A.; Sarma, D.; Papadas, I.T.; Armatas, G.S.; Hassan, Y.S.; Alkordi, M.H.; Kanatzidis, M.G.; Manos, M.J. Rapid, Green and Inexpensive Synthesis of High Quality UiO-66 Amino-Functionalized Materials with Exceptional Capability for Removal of Hexavalent Chromium from Industrial Waste. Inorg. Chem. Front. 2016, 3, 635–644. [Google Scholar] [CrossRef]
- Kot-Wasik, A.; Zabiegała, B.; Urbanowicz, M.; Dominiak, E.; Wasik, A.; Namieśnik, J. Advances in Passive Sampling in Environmental Studies. Anal. Chim. Acta 2007, 602, 141–163. [Google Scholar] [CrossRef]
- Roll, I.B.; Halden, R.U. Critical Review of Factors Governing Data Quality of Integrative Samplers Employed in Environmental Water Monitoring. Water Res. 2016, 94, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Kaserzon, S.L.; Kennedy, K.; Hawker, D.W.; Thompson, J.; Carter, S.; Roach, A.C.; Booij, K.; Mueller, J.F. Development and Calibration of a Passive Sampler for Perfluorinated Alkyl Carboxylates and Sulfonates in Water. Environ. Sci. Technol. 2012, 46, 4985–4993. [Google Scholar] [CrossRef]
- Męczykowska, H.; Kobylis, P.; Stepnowski, P.; Caban, M. Calibration of Passive Samplers for the Monitoring of Pharmaceuticals in Water-Sampling Rate Variation. Crit. Rev. Anal. Chem. 2017, 47, 204–222. [Google Scholar] [CrossRef]
- Morin, N.; Miège, C.; Coquery, M.; Randon, J. Chemical Calibration, Performance, Validation and Applications of the Polar Organic Chemical Integrative Sampler (POCIS) in Aquatic Environments. TrAC Trends Anal. Chem. 2012, 36, 144–175. [Google Scholar] [CrossRef] [Green Version]
- Harman, C.; Allan, I.J.; Vermeirssen, E.L.M. Calibration and Use of the Polar Organic Chemical Integrative Sampler—A Critical Review. Environ. Toxicol. Chem. 2012, 31, 2724–2738. [Google Scholar] [CrossRef]
- Arditsoglou, A.; Voutsa, D. Passive Sampling of Selected Endocrine Disrupting Compounds Using Polar Organic Chemical Integrative Samplers. Environ. Pollut. 2008, 156, 316–324. [Google Scholar] [CrossRef]
- Mechelke, J.; Vermeirssen, E.L.M.; Hollender, J. Passive sampling of organic contaminants across the water-sediment interface of an urban stream. Water Res. 2019, 165, 114966. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Xu, D.; Wang, Y.; Wang, Y.; Li, Y.; Gong, M.; Zhang, C. Simultaneous Measurements of Eight Oxyanions Using High-Capacity Diffusive Gradients in Thin Films (Zr-Oxide DGT) with a High-Efficiency Elution Procedure. Environ. Sci. Technol. 2016, 50, 7572–7580. [Google Scholar] [CrossRef]
- Guan, D.-X.; Williams, P.N.; Luo, J.; Zheng, J.-L.; Xu, H.-C.; Cai, C.; Ma, L.Q. Novel Precipitated Zirconia-Based DGT Technique for High-Resolution Imaging of Oxyanions in Waters and Sediments. Environ. Sci. Technol. 2015, 49, 3653–3661. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, H.; Santner, J.; Davison, W. Performance Characteristics of Diffusive Gradients in Thin Films Equipped with a Binding Gel Layer Containing Precipitated Ferrihydrite for Measuring Arsenic(V), Selenium(VI), Vanadium(V), and Antimony(V). Anal. Chem. 2010, 82, 8903–8909. [Google Scholar] [CrossRef]
- Bennett, W.W.; Teasdale, P.R.; Panther, J.G.; Welsh, D.T.; Jolley, D.F. Speciation of Dissolved Inorganic Arsenic by Diffusive Gradients in Thin Films: Selective Binding of As III by 3-Mercaptopropyl-Functionalized Silica Gel. Anal. Chem. 2011, 83, 8293–8299. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mixing Speed (rpm) a | Mass Sorbed (μg) b | Mass Sorbed per Mass of Sorbent (mg/g) | Mass Sorbed per Surface Area (mg/m) | Sampling Rate (L/d) | Sampling Rate per Surface Area (L/d × m2) | Linear Uptake Time (d) |
|---|---|---|---|---|---|---|---|
| As(V) | |||||||
| River water | 50 | 349 | 16.6 | 278 | 0.306 | 244 | 6 |
| 150 | 412 | 19.6 | 328 | 0.286 | 228 | 7 | |
| Lake water | 50 | 301 | 14.3 | 240 | 0.138 | 110 | 10 |
| 150 | 287 | 13.7 | 228 | 0.110 | 88 | 8 | |
| Se(IV) | |||||||
| River water | 50 | 537 | 25.6 | 427 | 0.296 | 236 | 7 |
| 150 | 415 | 19.8 | 330 | 0.324 | 258 | 6 | |
| Lake water | 50 | 264 | 12.6 | 210 | 0.110 | 88 | 10 |
| 150 | 760 | 36.2 | 605 | 0.323 | 257 | 10 |
| Sorbent | Sorption Capacity (mg/m2) | Reference | |
|---|---|---|---|
| As(V) a | Se(IV) a | ||
| Zirconium oxide gel | 27 (477) | 27 (240) | [33] |
| Precipitated Zr-oxide | 2.7 (426) | − (191) | [34] |
| Slurry ferrihydrite | >3.1 (100) | − (70) | [35,36] |
| Precipitated ferrihydrite | >3.7 (277) | − (118) | [35] |
| TiO2 powder | >3.0 (261) | − | [36] |
| MOR−1@cotton fabric | 228–328 (−) b | 210–605 (−) | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moisiadis, E.; Pournara, A.D.; Manos, M.J.; Giokas, D.L. Development of a New Method to Estimate the Water Purification Efficiency of Bulk-Supported Nanosorbents under Realistic Conditions. Separations 2023, 10, 140. https://doi.org/10.3390/separations10020140
Moisiadis E, Pournara AD, Manos MJ, Giokas DL. Development of a New Method to Estimate the Water Purification Efficiency of Bulk-Supported Nanosorbents under Realistic Conditions. Separations. 2023; 10(2):140. https://doi.org/10.3390/separations10020140
Chicago/Turabian StyleMoisiadis, Elias, Anastasia D. Pournara, Manolis J. Manos, and Dimosthenis L. Giokas. 2023. "Development of a New Method to Estimate the Water Purification Efficiency of Bulk-Supported Nanosorbents under Realistic Conditions" Separations 10, no. 2: 140. https://doi.org/10.3390/separations10020140
APA StyleMoisiadis, E., Pournara, A. D., Manos, M. J., & Giokas, D. L. (2023). Development of a New Method to Estimate the Water Purification Efficiency of Bulk-Supported Nanosorbents under Realistic Conditions. Separations, 10(2), 140. https://doi.org/10.3390/separations10020140