


Optimization of Chromatographic Conditions with QbD for Method Development and Validation of Bosutinib by HPLC: Applications in Dosage Forms and Rat Plasma Analysis

 ,
,  ,
,
Abstract
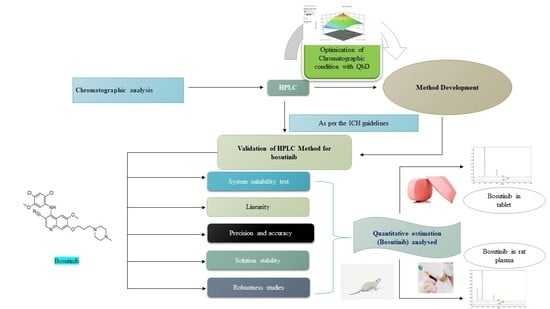
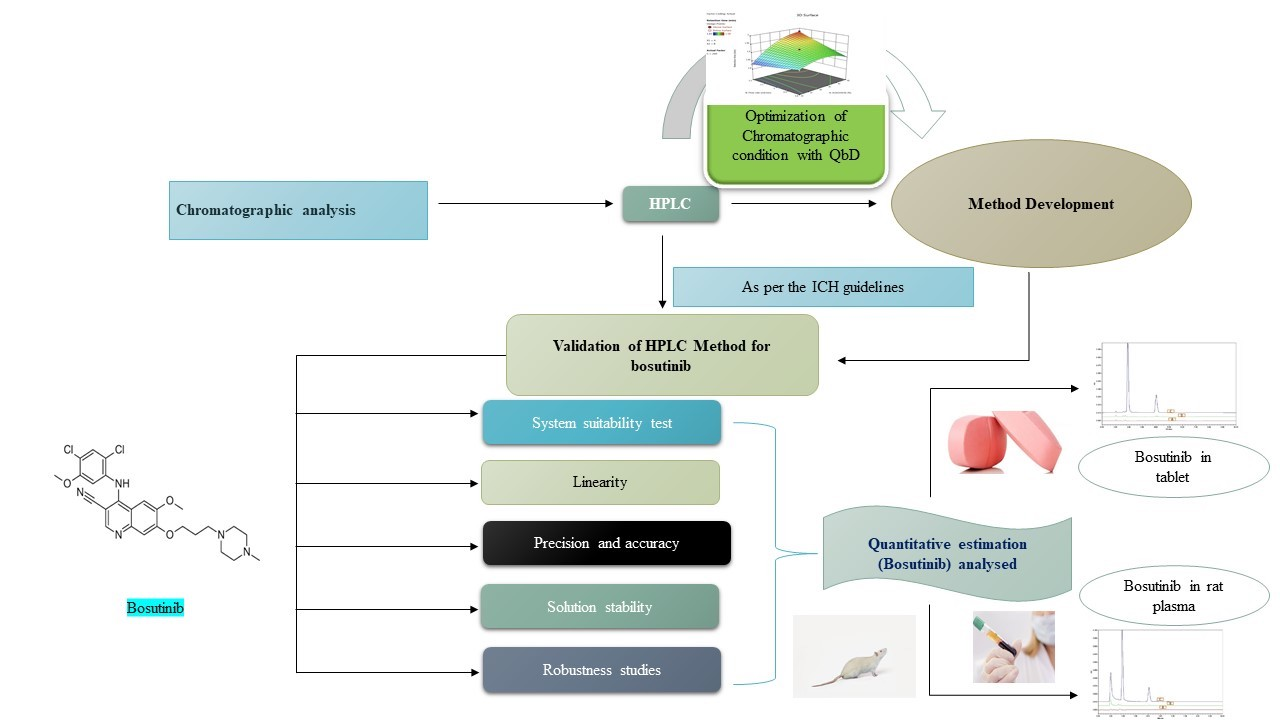
:
1. Introduction
2. Experimental Work
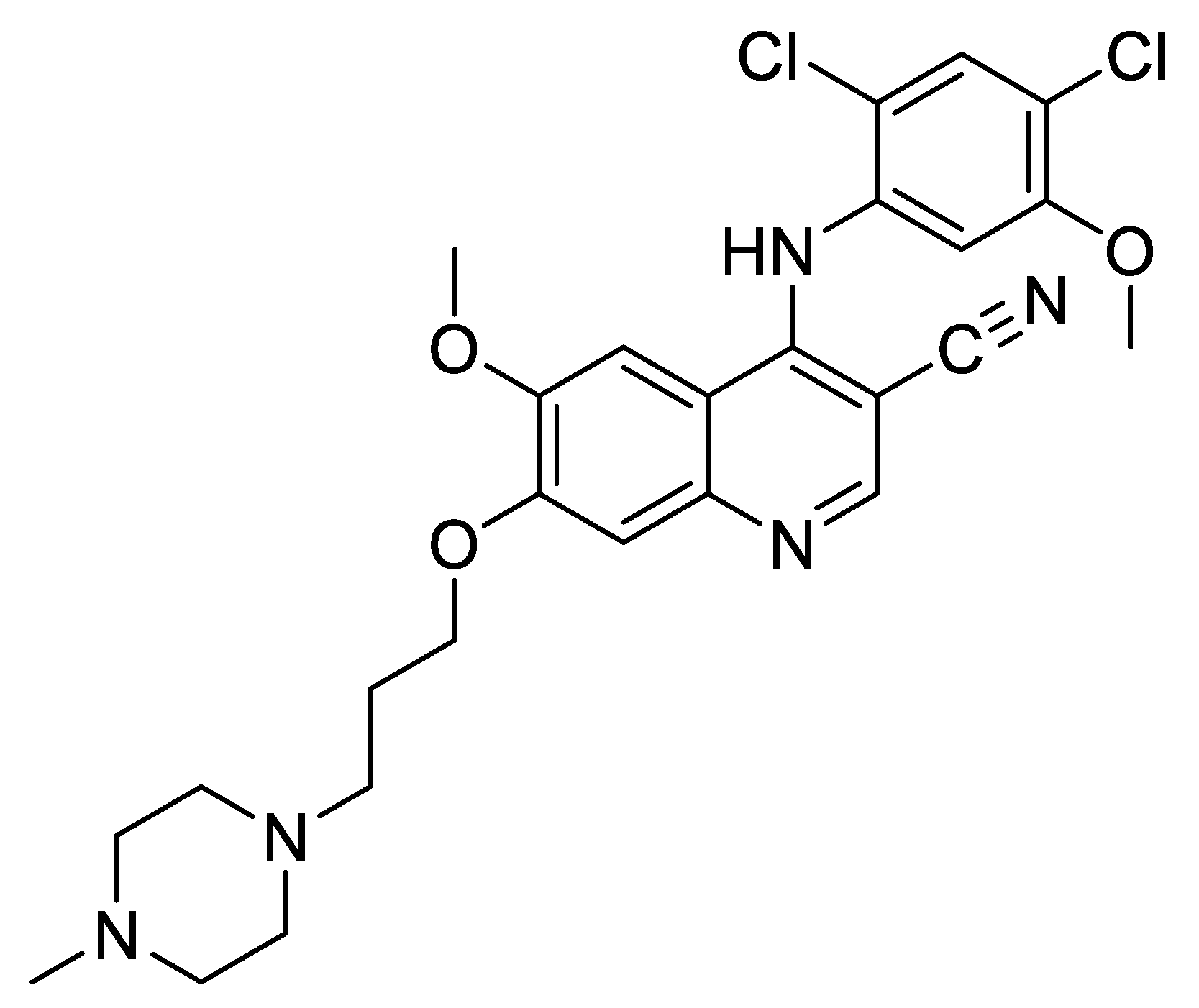
2.1. Materials
2.2. Instruments/Apparatus
2.3. Methods
2.3.1. Risk Assessment Studies
2.3.2. Optimization
2.3.3. Method Development
2.3.4. Stock, Standard, and Quality-Control Sample
2.3.5. Preparation of Calibration Curve
2.3.6. Preparation of Tablet Formulation Solution
2.3.7. Preparation of Rat Plasma Samples
2.4. Method Validation
2.4.1. System Suitability Test
2.4.2. Linearity
2.4.3. Precision and Accuracy
2.4.4. Solution Stability
2.4.5. Robustness Study
3. Results and Discussion
3.1. Risk Assessment Studies
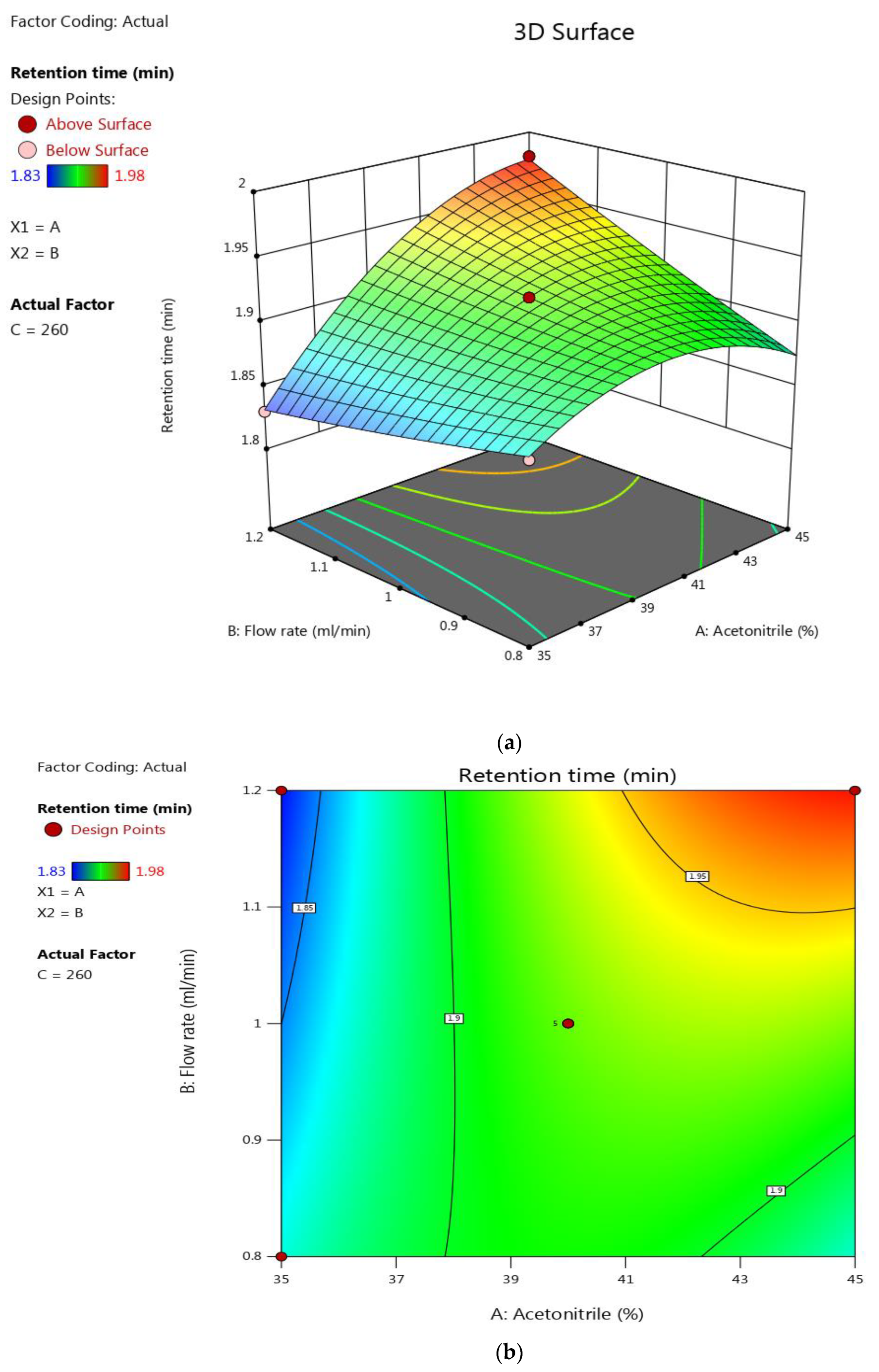
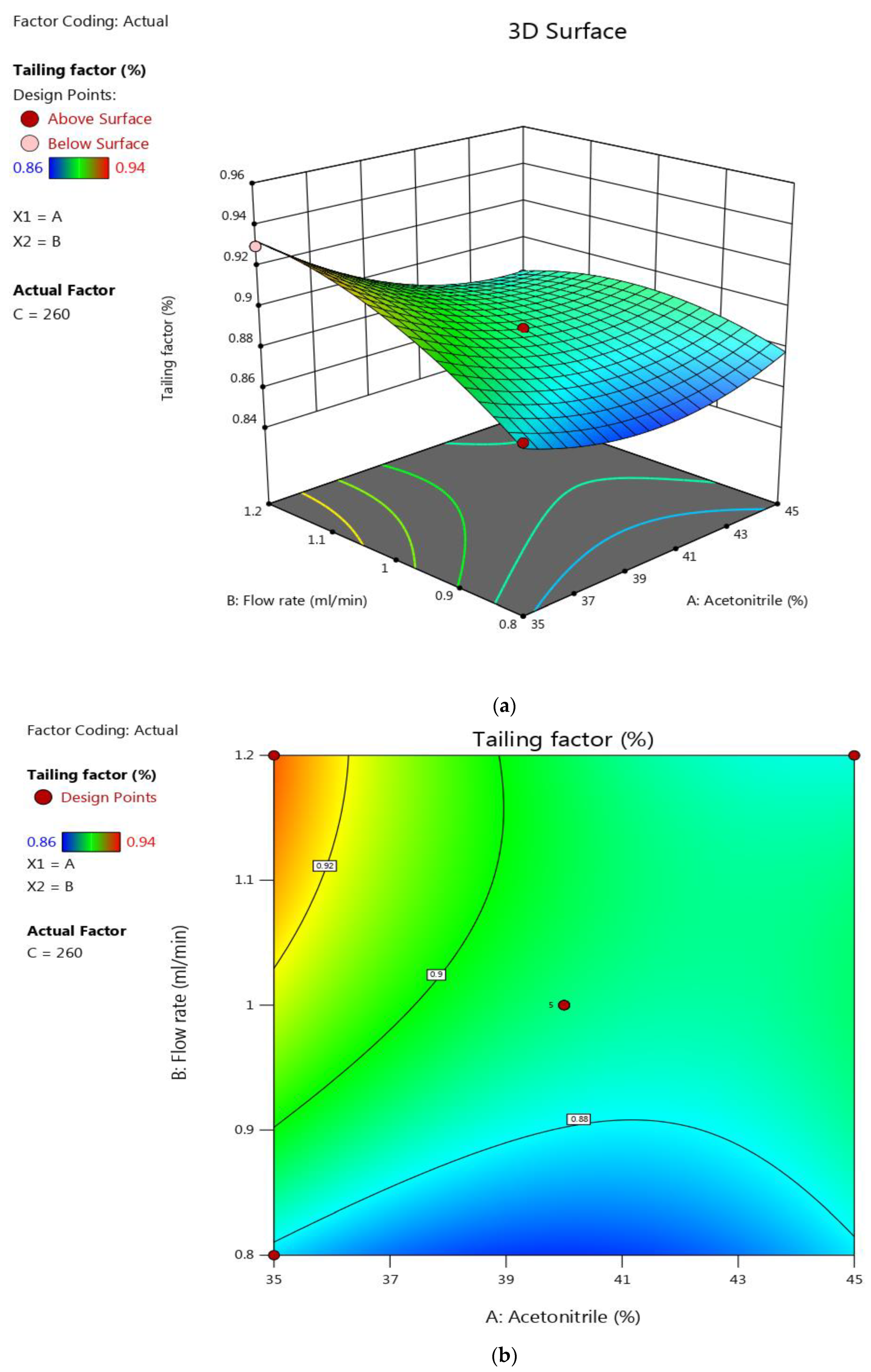
3.2. Optimization
3.3. Stock, Standard and Quality Control Sample
3.4. Method Validation
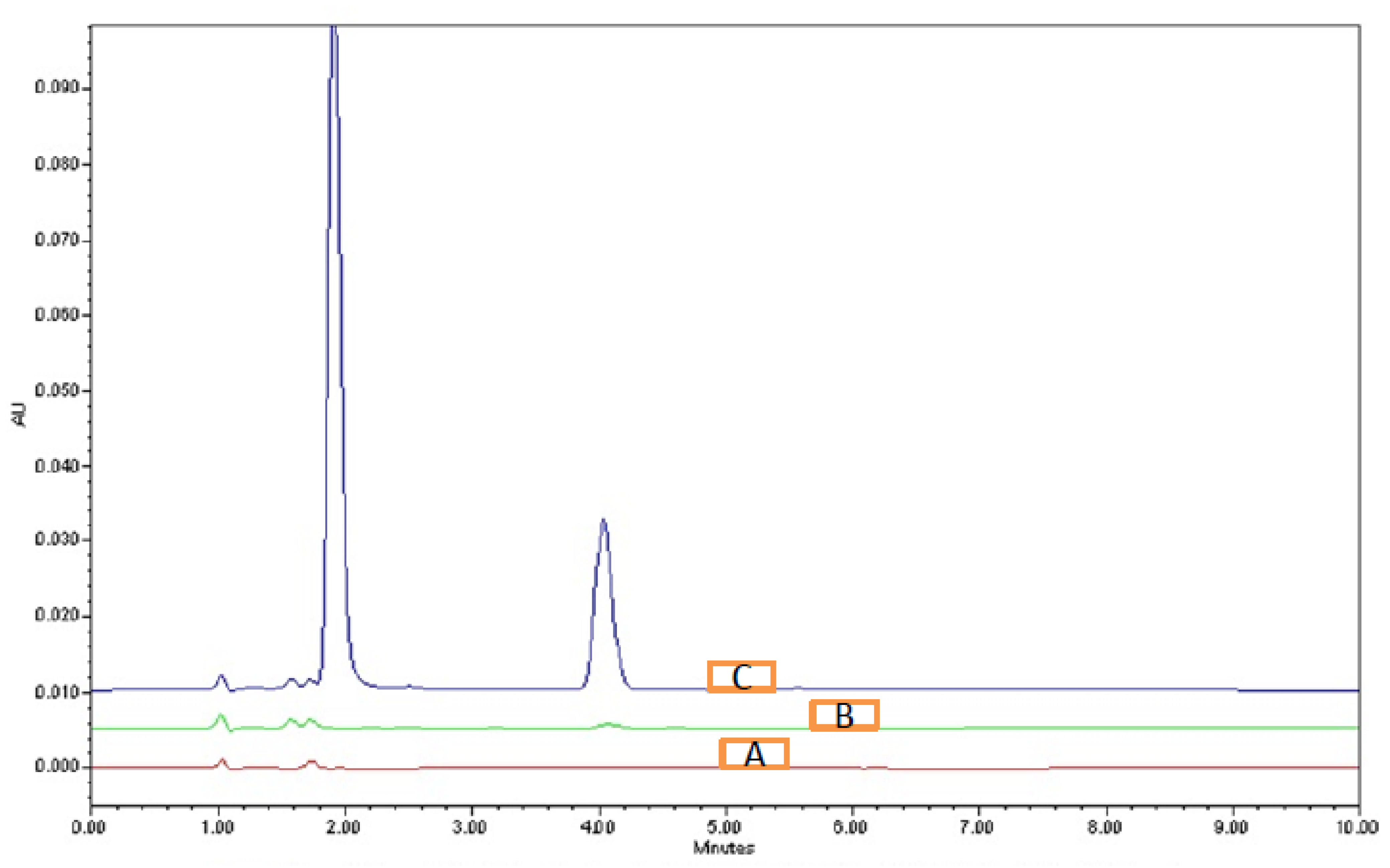
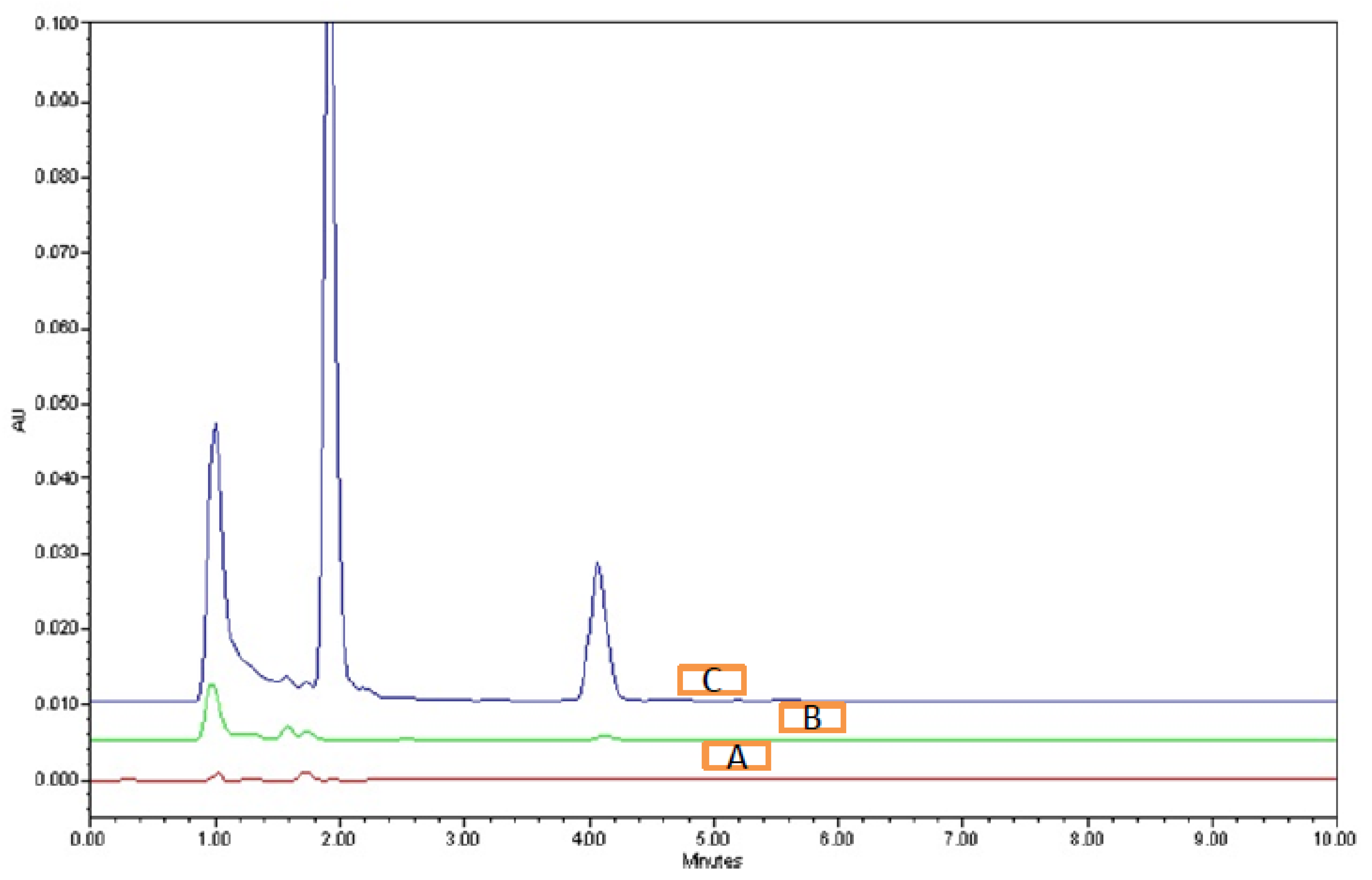
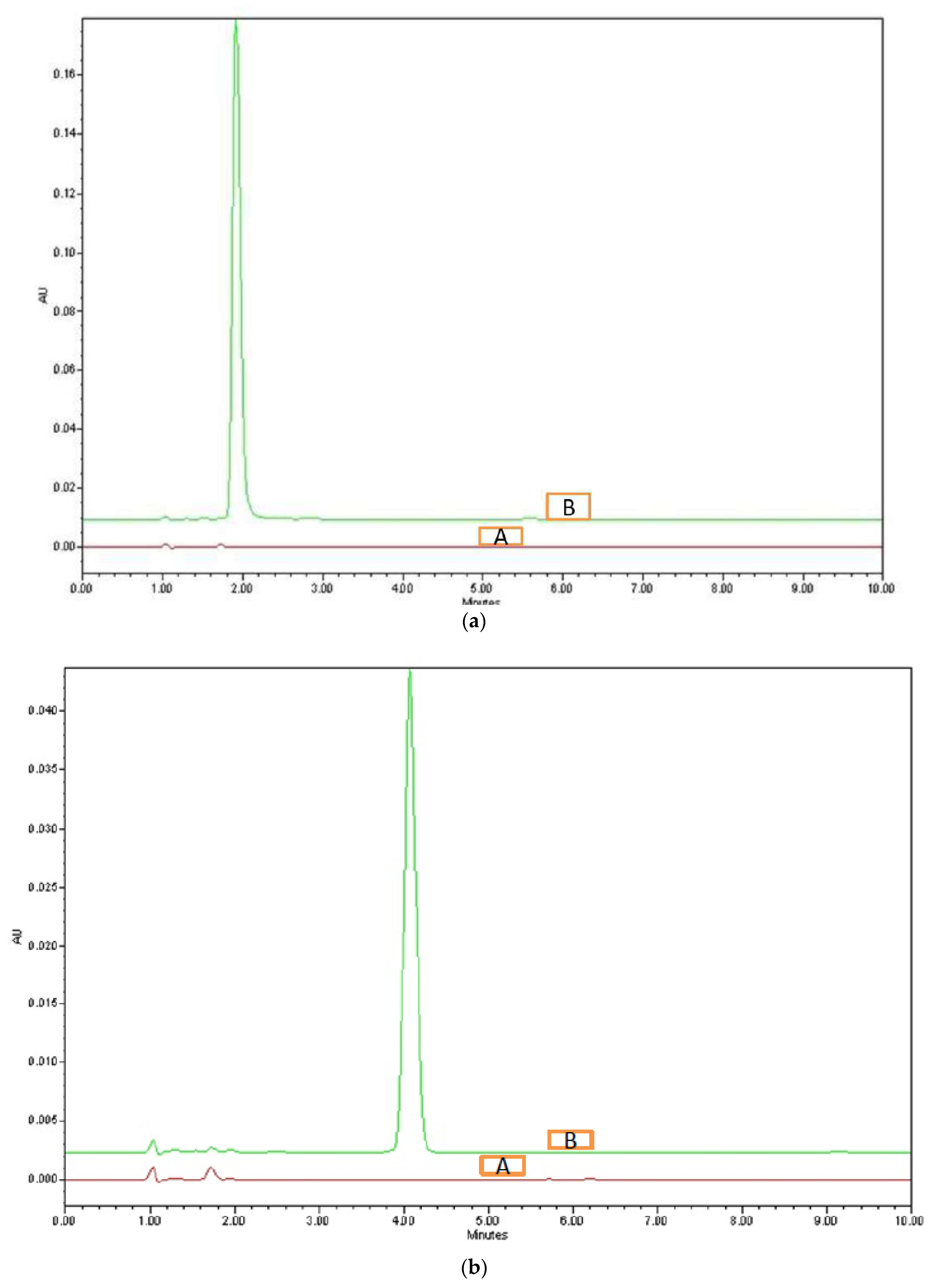
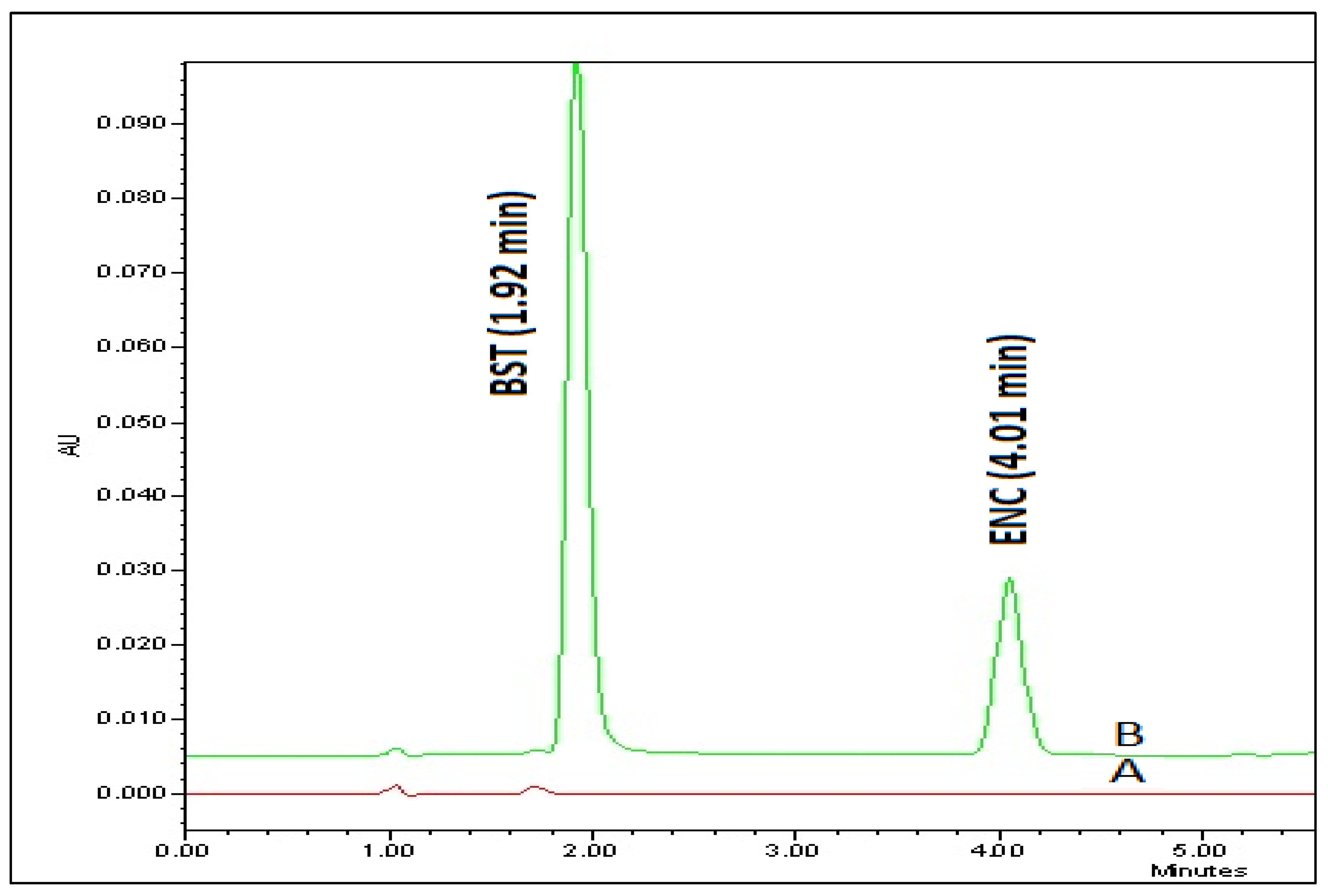
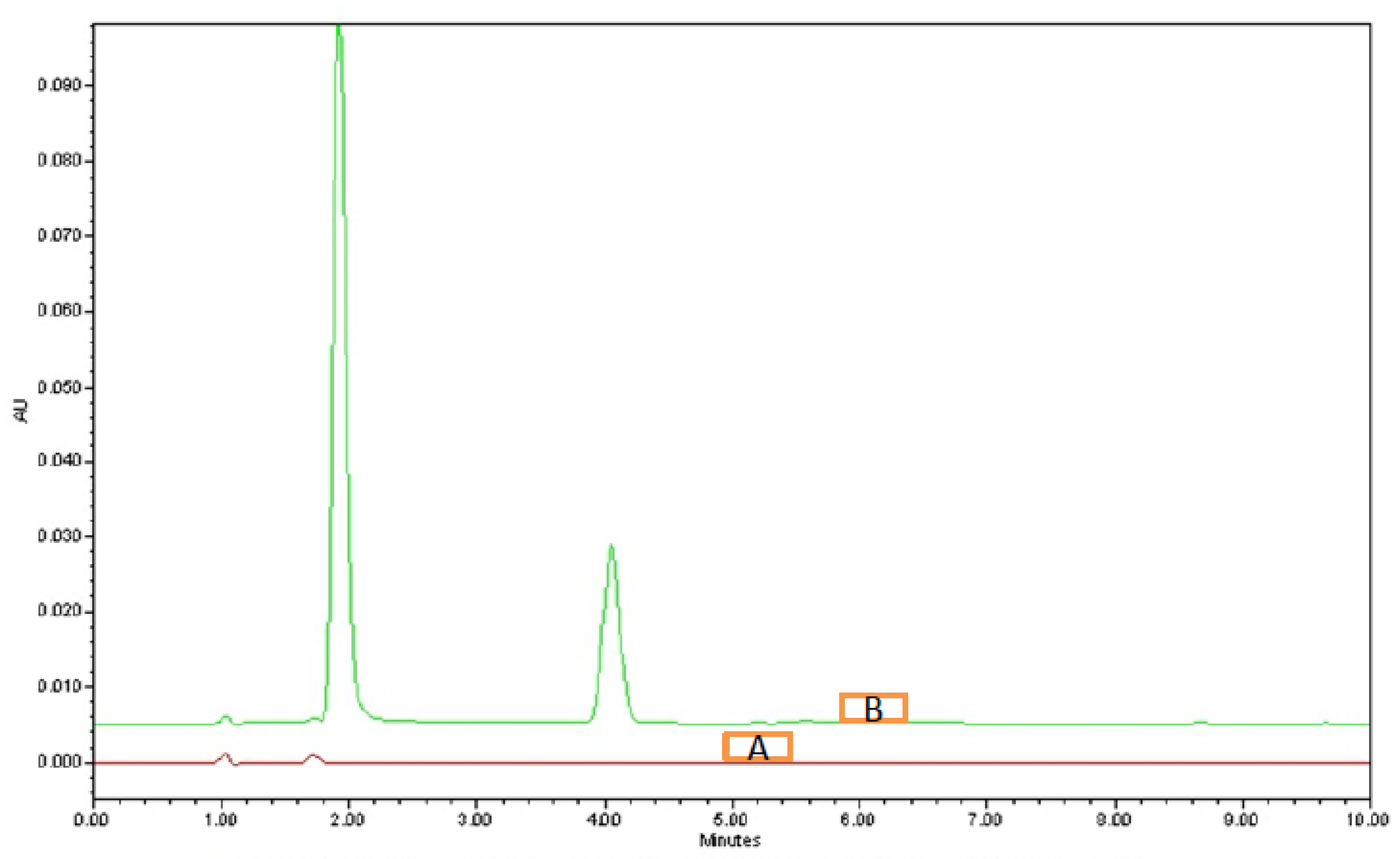
3.4.1. System Suitability
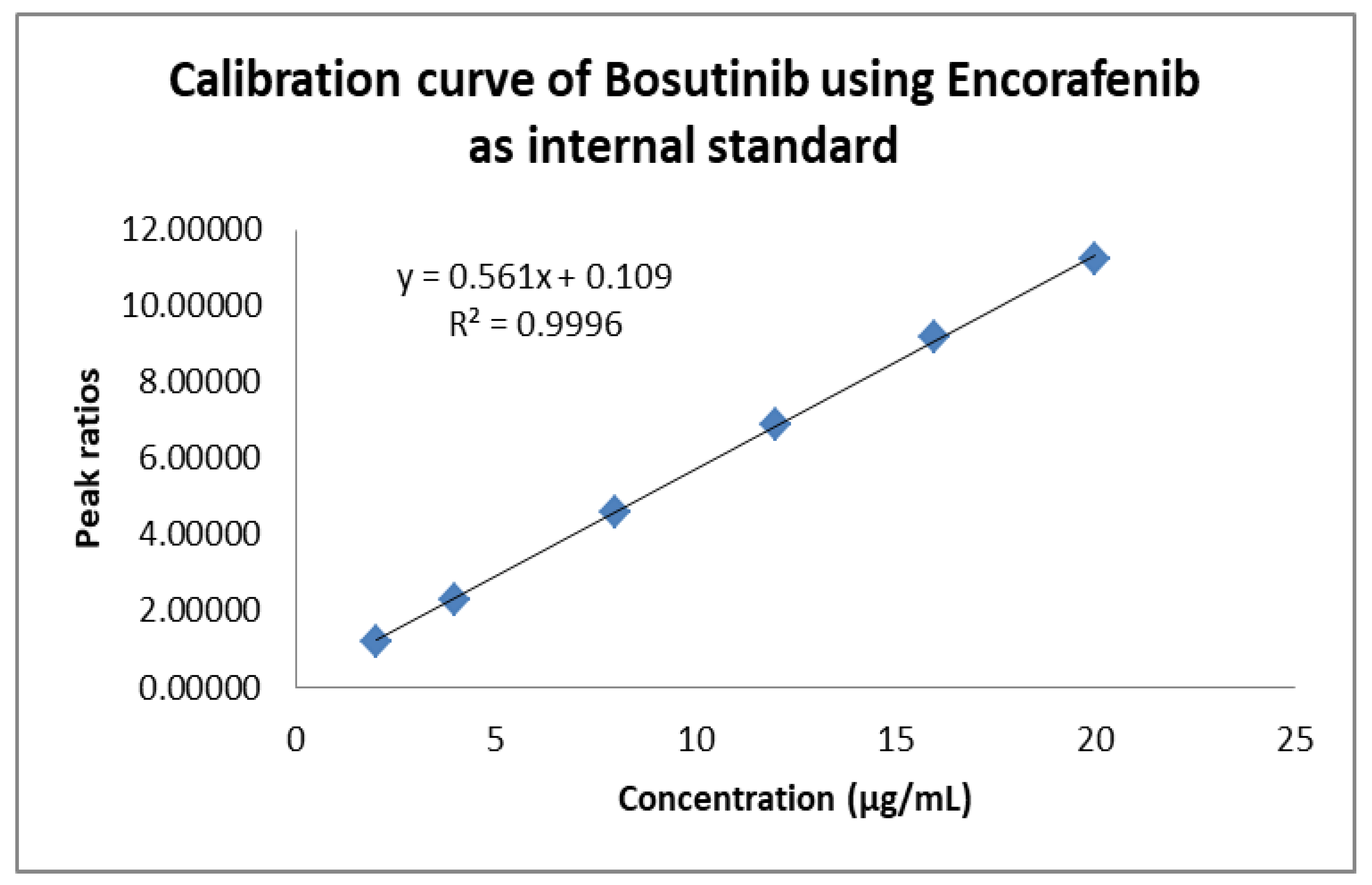
3.4.2. Linearity
3.4.3. Precision and Accuracy
3.4.4. Solution Stability
3.4.5. Robustness Study
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbas, R.; Hsyu, P.H. Clinical Pharmacokinetics and Pharmacodynamics of Bosutinib. Clin. Pharmacokinet. 2016, 55, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Haap, M.; Kopp, H.G.; Lipp, H.P. Tyrosine kinase inhibitors—A review on pharmacology, metabolism and side effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.; Schafhausen, P.; Brummendorf, T.H. Bosutinib: A dual SRC/ABL kinase inhibitor for the treatment of chronic myeloid leukemia. Expert. Rev. Hematol. 2009, 2, 489–497. [Google Scholar] [CrossRef]
- Smith, J.H.; McNair, H.M. Fast HPLC with a silica-based monolithic ODS column. J. Chromatogr. Sci. 2003, 41, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef] [Green Version]
- Dziadosz, M.; Lessig, R.; Bartels, H. HPLC-DAD protein kinase inhibitor analysis in human serum. J. Chromatogr. B 2012, 893, 77–81. [Google Scholar] [CrossRef]
- Sumimoto, T.; Nakahara, R.; Sato, Y.; Itoh, H. A quantitative method for the determination of bosutinib in human plasma using high-performance liquid chromatography and ultraviolet detection. J. Clin. Lab. Anal. 2018, 32, e22201. [Google Scholar] [CrossRef] [Green Version]
- Mita, A.; Abumiya, M.; Miura, M.; Niioka, T.; Takahashi, S.; Yoshioka, T.; Kameoka, T.; Takahashi, N. Correlation of plasma concentration and adverse effects of bosutinib: Standard dose or dose-escalation regimens of bosutinib treatment for patients with chronic myeloid leukemia. Exp. Hematol. Oncol. 2018, 7, 9. [Google Scholar] [CrossRef]
- Wang, L.; Tang, L.; Zheng, Y.; Pan, G.; Zhu, W.; Pan, C.; Zhu, L. Determination of bosutinib in mice plasma and tissue by UPLC-MS/MS and its application to the pharmacokinetic and tissue distribution study. Anal. Method 2015, 7, 9184–9189. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, X.C.; Dai, S.; Xiao, Y.; Zhou, M.T. A simple method for the determination of Bosutinib in rat plasma by UPLC-MS/MS. J. Chromatogr. B 2015, 1004, 93–97. [Google Scholar] [CrossRef]
- Jadhav, P.B.; Gajare, G.K. Development and Validation of an RP-HPLC method for bosutinib in bulk dosage form. Int. J. Res. Pharm. Chem. 2016, 6, 599–603. [Google Scholar]
- Prajapati, P.B.; Bagul, N.; Kalyankar, G. Implementation of DoE and Risk-Based Enhanced Analytical Quality by Design Approach to Stability-Indicating RP-HPLC Method for Stability Study of Bosutinib. J. AOAC Int. 2021, 104, 1742–1753. [Google Scholar] [CrossRef]
- Raman, N.V.; Mallu, U.R.; Bapatu, H.R. Analytical Quality by Design Approach to Test Method Development and Validation in Drug Substance Manufacturing. J. Chem. 2015, 2015, 435129. (Pages-8). [Google Scholar] [CrossRef]
- Konda, R.K.; Challa, B.R.; Chandu, B.R.; Chandrasekhar, K.B. Bioanalytical method development and validation of memantine in human plasma by high performance liquid chromatography with tandem mass spectrometry: Application to bioequivalence study. J. Anal. Methods Chem. 2012, 2012, 101249. (Pages-8). [Google Scholar] [CrossRef] [Green Version]
- ICH, Geneva (Switzerland). Q2 (R1) Validation of Analytical Procedures: Text and Methodology 1994/1996. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 5 March 2022).
- USFDA USA, Centre for Drug Evaluation and Research (CDER). Reviewer Guidance: Validation of Chromatographic Methods, Rockville 1994. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/reviewer-guidance-validation-chromatographic-methods (accessed on 5 April 2022).
- USFDA USA. Guidance for Industry: Analytical Procedures and Methods Validation; Chemistry Manufacturing and Control Documentation, Rockville 2000. Available online: https://www.astrixinc.com/data/integrity (accessed on 5 April 2022).
- Jain, P.; Taleuzzaman, M.; Kala, C.; Kumar Gupta, D.; Ali, A.; Aslam, M. Quality by design (Qbd) assisted development of phytosomal gel of aloe vera extract for topical delivery. J. Liposome Res. 2021, 31, 381–388. [Google Scholar] [CrossRef]
- Panda, S.S.; Ravi Kumar Bera, V.V.; Beg, S.; Mandal, O. Analytical Quality by Design (AQbD)-Oriented RP-UFLC Method for Quantification of Lansoprazole with Superior Method Robustness. J. Liq. Chromatogr. Relat. Technol. 2017, 40, 479–485. [Google Scholar] [CrossRef]
- Peraman, R.; Bhadraya, K.; Reddy, Y.P.; Reddy, C.S.; Lokesh, T. Analytical Quality by Design Approach in RP-HPLC Method Development for the Assay of Etofenamate in Dosage Forms. Ind. J. Pharm. Sci. 2015, 77, 751–757. [Google Scholar] [CrossRef]
- Ameeduzzafar El-Bagory, I.; Alruwaili, N.K.; Imam, S.S.; Alomar, F.A.; Elkomy, M.H.; Ahmad, N.; Elmowafy, M. Quality by design (QbD) based development and validation of bioanalytical RP-HPLC method for dapagliflozin: Forced degradation and preclinical pharmacokinetic study. J. Liq. Chromatogr. Relat. Technol. 2020, 43, 53–65. [Google Scholar] [CrossRef]
- Prajapati, P.B.; Patel, A.S.; Shah, S.A. DoE-Based Analytical-FMCEA for Enhanced AQbD Approach to MEER-RP-HPLC Method for Synchronous Estimation of Fifteen Antihypertensive Pharmaceutical Dosage Forms. J. AOAC Int. 2022, 105, 34–45. [Google Scholar] [CrossRef]
- Mohanty, D.; Rani, M.J.; Haque, M.A.; Bakshi, V.; Jahangir, M.A.; Imam, S.S.; Gilani, S.J. Preparation and evaluation of transdermal naproxen niosomes: Formulation optimization to preclinical anti-inflammatory assessment on murine model. J. Liposome Res. 2020, 30, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Rahat, I.; Imam, S.S.; Rizwanullah, M.; Alshehri, S.; Asif, M.; Kala, C.; Taleuzzamane, M. Thymoquinone-entrapped chitosan-modified nanoparticles: Formulation optimization to preclinical bioavailability assessments. Drug Deliv. 2021, 28, 973–984. [Google Scholar] [CrossRef]
- International Conference on Harmonization (ICH: Q2). Guideline on Validation of Analytical Procedures: Text and Methodology 2005. Available online: http://www.ich.org/products/guidelines/quality/quality-single/article/validation-of-analytical-procedurestext-and-methodology.html (accessed on 21 July 2014).
- The United States Pharmacopoeia 35 National Formulary 30, Eas-Ton; Rand Mc Nally Taunton: Chicago, IL, USA, 2012.
- Gillings, N.; Todde, S.; Behe, M.; Decristoforo, C.; Elsinga, P.; Ferrari, V.; Patt, M. EANM guideline on the validation of analytical methods for radiopharmaceuticals. EJNMMI Radiopharm. Chem. 2020, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Amer, S.M.; Kadi, A.A.; Darwish, H.W.; Attwa, M.W. LC-MS/MS method for the quantification of masitinib in RLMs matrix and rat urine: Application to metabolic stability and excretion rate. Chem. Cent. J. 2017, 11, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadi, A.A.; Darwish, H.W.; Attwa, M.W.; Amer, S.M. Validated LC-MS/MS Method for the Quantification of Ponatinib in Plasma: Application to Metabolic Stability. PLoS ONE 2016, 11, e0164967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabhi, B.; Jadeja, Y.; Patel, M.; Jebaliya, H.; Karia, D.; Shah, A. Method Development and Validation of a Stability-Indicating RP-HPLC Method for the Quantitative Analysis of Dronedarone Hydrochloride in Pharmaceutical Tablets. Sci. Pharm. 2013, 81, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Tome, T.; Žigart, N.; Časar, Z.; Obreza, A. Development and optimization of liquid chromatography analytical methods by using AQbD principles: Overview and recent advances. Org. Process Res. Dev. 2019, 23, 1784–1802. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concn.(μg/mL) | Standard Stock Solution (mL) (BST) (μg/mL) | Internal Standard (mL) (ENC) (100 μg/mL) | Mobile Phase (mL) (A:B 60:40 v/v) | Total Volume (mL) |
|---|---|---|---|---|
| 8 μg/mL (80%) | 0.16 | 0.4 | 1.44 | 2.0 |
| 10 μg/mL (100%) | 0.20 | 0.4 | 1.40 | 2.0 |
| 12 μg/mL (120%) | 0.24 | 0.4 | 1.36 | 2.0 |
| Bosutinib Chromatographic Technique Features | |||||||
|---|---|---|---|---|---|---|---|
| CAAs | Eluent System Components | Wavelength of the Sensor | Column Temperature | Sample Volume | Flow Speed | Flow Pattern | Column Sizes |
| Retention time | +1 | 0 | 0 | 0 | +1 | 0 | 0 |
| Peak Area | +1 | 0 | −1 | 0 | +1 | −1 | 0 |
| Tailing Factor | +1 | −1 | −1 | 0 | 0 | 0 | 0 |
| S. No. | Independent Variable | Dependent Variable | |||
|---|---|---|---|---|---|
| Standard Run | Acetonitrile (%) | Flow Speed (mL/min) | Wavelength (nm) | Retention Time (min) | Tailing Factor |
| 1. | 35 | 0.8 | 260 | 1.87 | 0.88 |
| 2. | 45 | 0.8 | 255 | 1.89 | 0.87 |
| 3. | 35 | 1.2 | 260 | 1.83 | 0.93 |
| 4. | 45 | 1.2 | 260 | 1.98 | 0.88 |
| 5. | 35 | 1 | 250 | 1.92 | 0.94 |
| 6. | 45 | 1 | 250 | 1.93 | 0.88 |
| 7. | 35 | 1 | 270 | 1.84 | 0.9 |
| 8. | 45 | 1 | 270 | 1.97 | 0.91 |
| 9. | 40 | 0.8 | 250 | 1.98 | 0.86 |
| 10. | 40 | 1.2 | 250 | 1.95 | 0.92 |
| 11. | 40 | 0.8 | 270 | 1.89 | 0.87 |
| 12. | 40 | 1.2 | 270 | 1.98 | 0.88 |
| 13 *. | 40 | 1 | 260 | 1.92 | 0.89 |
| 14 *. | 40 | 1 | 260 | 1.92 | 0.89 |
| 15 *. | 40 | 1 | 260 | 1.92 | 0.89 |
| 16 *. | 40 | 1 | 260 | 1.92 | 0.89 |
| 17 *. | 40 | 1 | 260 | 1.92 | 0.89 |
| Retention Time (min) | Tailing Factor | Plate Count | ||||
|---|---|---|---|---|---|---|
| BST | ENC | BST | ENC | BST | ENC | |
| 1 | 1.94 | 4.06 | 0.9 | 1.21 | 2032 | 4288 |
| 2 | 1.95 | 3.99 | 0.88 | 1.2 | 2018 | 4232 |
| 3 | 1.91 | 3.97 | 0.9 | 1.21 | 2013 | 4276 |
| 4 | 1.89 | 4.05 | 0.91 | 1.18 | 2012 | 4232 |
| 5 | 1.93 | 3.99 | 0.87 | 1.18 | 2022 | 4284 |
| 6 | 1.93 | 4.06 | 0.91 | 1.17 | 2003 | 4224 |
| Average | 1.925 | 4.02 | 0.895 | 1.19167 | 2016.667 | 4256 |
| SD | 0.0197906 | 0.03742 | 0.015 | 0.01572 | 9.012337 | 27.03085 |
| %RSD | 0.98 | 0.92 | 1.68 | 1.26 | 4.46 | 0.63 |
| S. No. | Parameters | Numerical Value |
|---|---|---|
| 1 | Linearity range (µg mL−1) | 2–20 |
| 2 | Intercept | 0.109 |
| 3 | Slope | 0.561 |
| 4 | Correlation coefficient (r2) | 0.999 |
| 5 | SD | 0.098 |
| 6 | %RSD | 1.659 |
| 7 | LOD (µg mL−1) | 0.496 |
| 8 | LOQ (µg mL−1) | 1.503 |
| Precision | ||||||
|---|---|---|---|---|---|---|
| Inter-Day | Intraday | |||||
| Theoretical concentration (µg mL−1) | Concentration found | %RSD | Theoretical concentration (µg mL−1) | Concentration found | %RSD | |
| LQC | 2 | 1.97 ± 0.037 | 1.892 | 2 | 2.08 ± 0.04 | 1.923 |
| MQC | 12 | 11.91 ± 0.222 | 1.874 | 12 | 11.96 ± 0.21 | 1.762 |
| HQC | 20 | 19.89 ± 0.342 | 1.723 | 20 | 19.91 ± 0.362 | 1.823 |
| Accuracy (% Recovery) (Rat Plasma Samples) | ||||||
| Excess drug added to analyte % | Theoretical Content (µg mL−1) | Concentration found (Mean ± SD) | % Recovery (Mean ± SD) | %RSD | ||
| 0 | 8 | 7.86 ± 0.213 | 98.25 ± 1.847 | 1.881 | ||
| 50 | 12 | 11.84 ± 0.517 | 98.66 ± 1.954 | 1.982 | ||
| 100 | 16 | 16.2 ± 0.673 | 101.25 ± 1.783 | 1.763 | ||
| 150 | 20 | 20.08 ± 0.382 | 100.4 ± 1.482 | 1.476 | ||
| At 25 °C for 14 Days | At 2–8 °C for 30 Days | ||||||
|---|---|---|---|---|---|---|---|
| A | B | C | %RSD | A | B | C | %RSD |
| 12 | 11.85 ± 0.223 | 98.75 ± 1.852 | 1.875 | 12 | 11.81 ± 0.186 | 98.41 ± 1.624 | 1.651 |
| Retention Time (min) | Tailing Factor | Plate Count | ||||
|---|---|---|---|---|---|---|
| BST | ENC | BST | ENC | BST | ENC | |
| Ammonium acetate buffer pH 3.1 (Mobile phase A) | 1.93 | 4.08 | 0.85 | 1.21 | 2089 | 4421 |
| Ammonium acetate buffer pH 2.9 (Mobile phase A) | 1.91 | 4.12 | 0.91 | 1.16 | 2098 | 4212 |
| Flow rate at 0.9 mL min−1 | 1.90 | 3.95 | 0.92 | 1.22 | 1988 | 4098 |
| Flow rate at 1.1 mL min−1 | 1.95 | 4.13 | 0.93 | 1.18 | 1997 | 4235 |
| Detector wavelength at 258 nm | 1.93 | 3.98 | 0.86 | 1.23 | 2095 | 4312 |
| Detector wavelength at 262 nm | 1.94 | 4.05 | 0.88 | 1.17 | 2084 | 4308 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najmi, A.; Rehman, Z.u.; Alhazmi, H.A.; Albratty, M.M.; Majrashi, N.H.; Hakami, K.M.; Najmi, N.A.; Mobarki, A.A. Optimization of Chromatographic Conditions with QbD for Method Development and Validation of Bosutinib by HPLC: Applications in Dosage Forms and Rat Plasma Analysis. Separations 2023, 10, 346. https://doi.org/10.3390/separations10060346
Najmi A, Rehman Zu, Alhazmi HA, Albratty MM, Majrashi NH, Hakami KM, Najmi NA, Mobarki AA. Optimization of Chromatographic Conditions with QbD for Method Development and Validation of Bosutinib by HPLC: Applications in Dosage Forms and Rat Plasma Analysis. Separations. 2023; 10(6):346. https://doi.org/10.3390/separations10060346
Chicago/Turabian StyleNajmi, Asim, Zia ur Rehman, Hassan Ahmed Alhazmi, Mohammed Mofarreh Albratty, Nasser Hassan Majrashi, Khalid Mohammed Hakami, Naif Ali Najmi, and Ammar Abdullah Mobarki. 2023. "Optimization of Chromatographic Conditions with QbD for Method Development and Validation of Bosutinib by HPLC: Applications in Dosage Forms and Rat Plasma Analysis" Separations 10, no. 6: 346. https://doi.org/10.3390/separations10060346
APA StyleNajmi, A., Rehman, Z. u., Alhazmi, H. A., Albratty, M. M., Majrashi, N. H., Hakami, K. M., Najmi, N. A., & Mobarki, A. A. (2023). Optimization of Chromatographic Conditions with QbD for Method Development and Validation of Bosutinib by HPLC: Applications in Dosage Forms and Rat Plasma Analysis. Separations, 10(6), 346. https://doi.org/10.3390/separations10060346