Determination of Synthetic Cathinones in Urine and Oral Fluid by Liquid Chromatography High-Resolution Mass Spectrometry and Low-Resolution Mass Spectrometry: A Method Comparison





Abstract
:1. Introduction
2. Materials and Methods
2.1. Standards and Materials
2.2. HPLC Separation
2.3. Orbitrap
2.4. QqQ
2.5. Urine Collection and Preparation
2.6. Saliva Collection and Preparation
2.7. Validation
3. Results and Discussion
3.1. Urine Samples
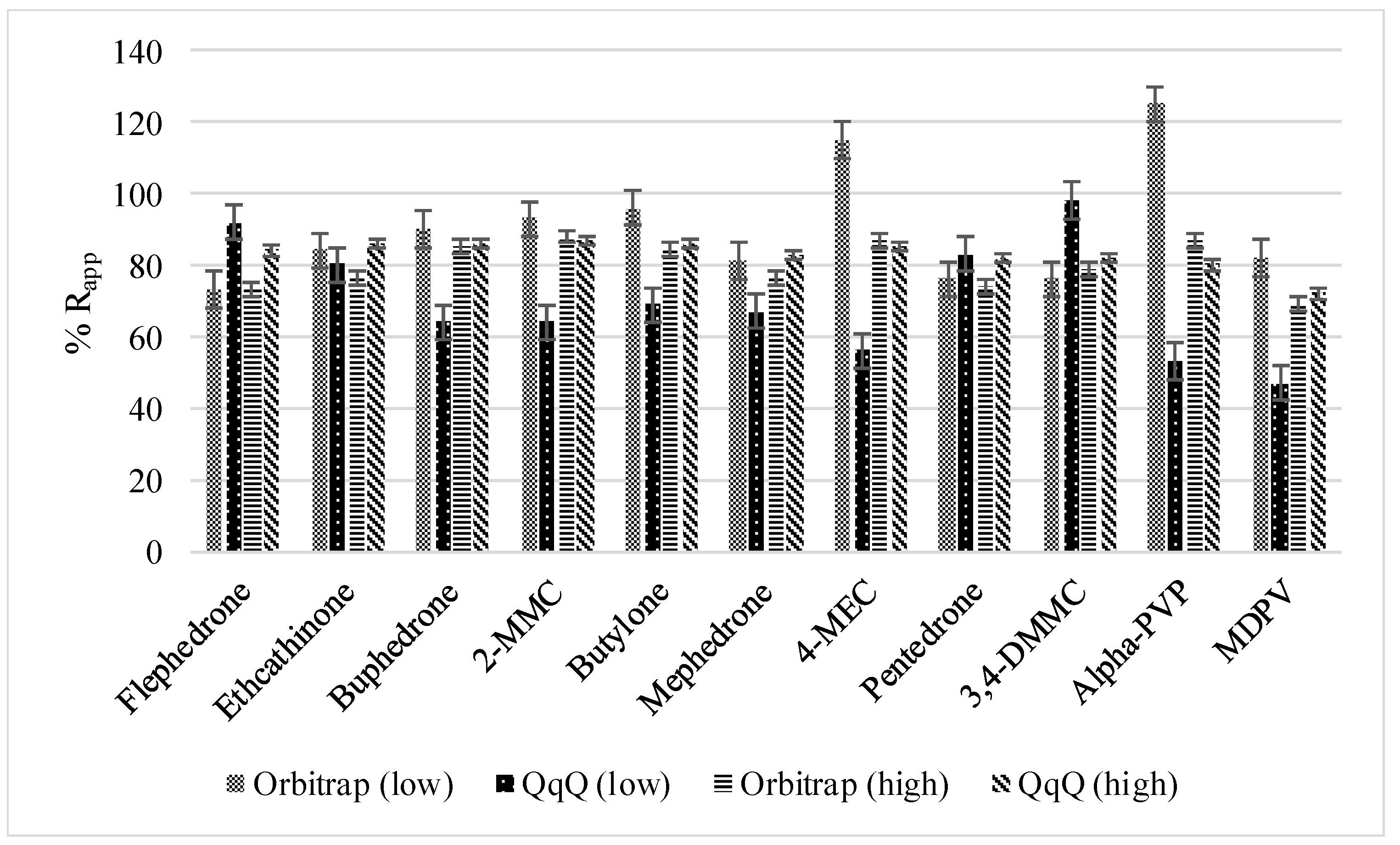
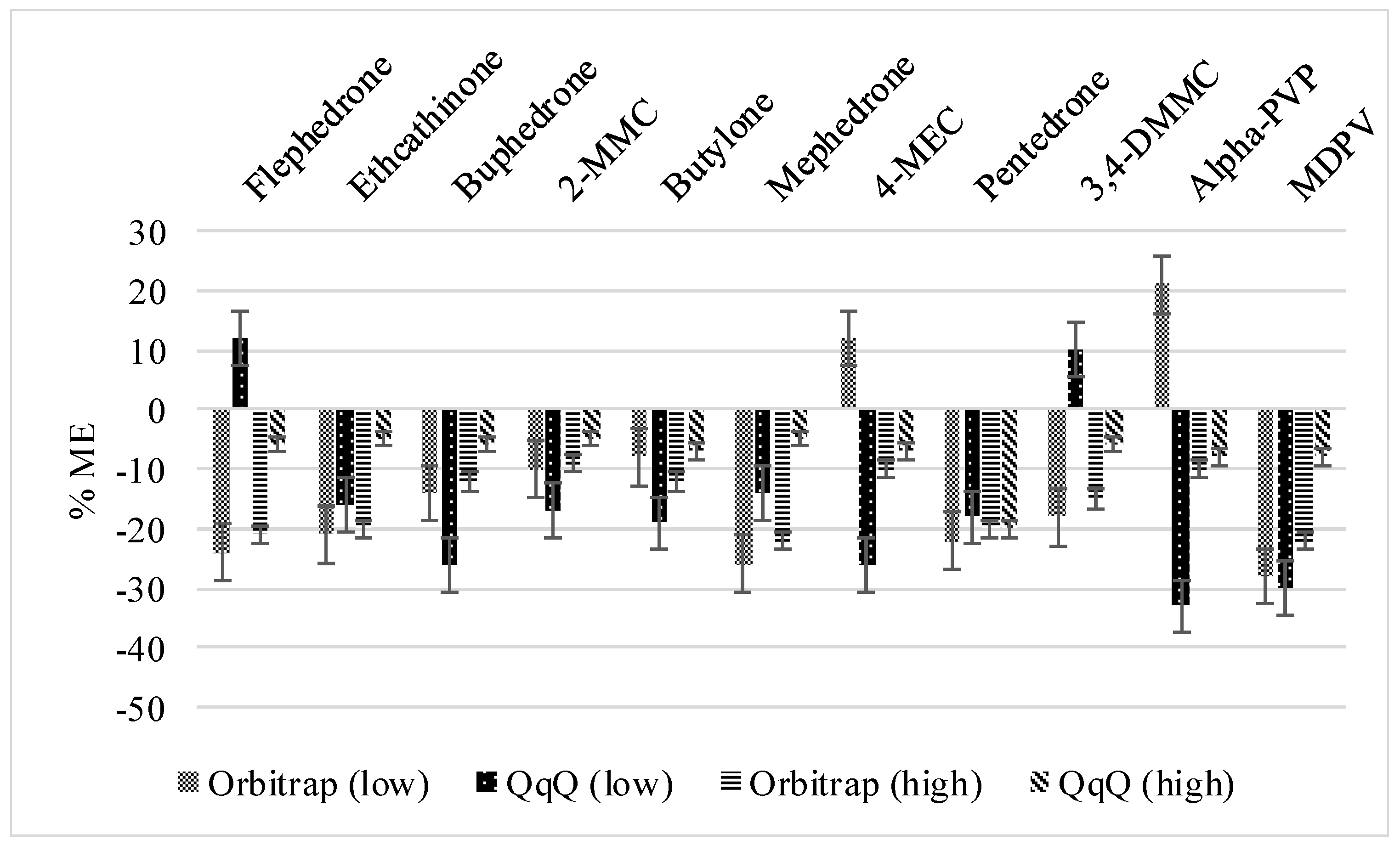
3.1.1. Method Recoveries and Matrix Effects
3.1.2. Linearity, MDLs, and MQLs
3.1.3. Accuracy
3.2. OF Samples
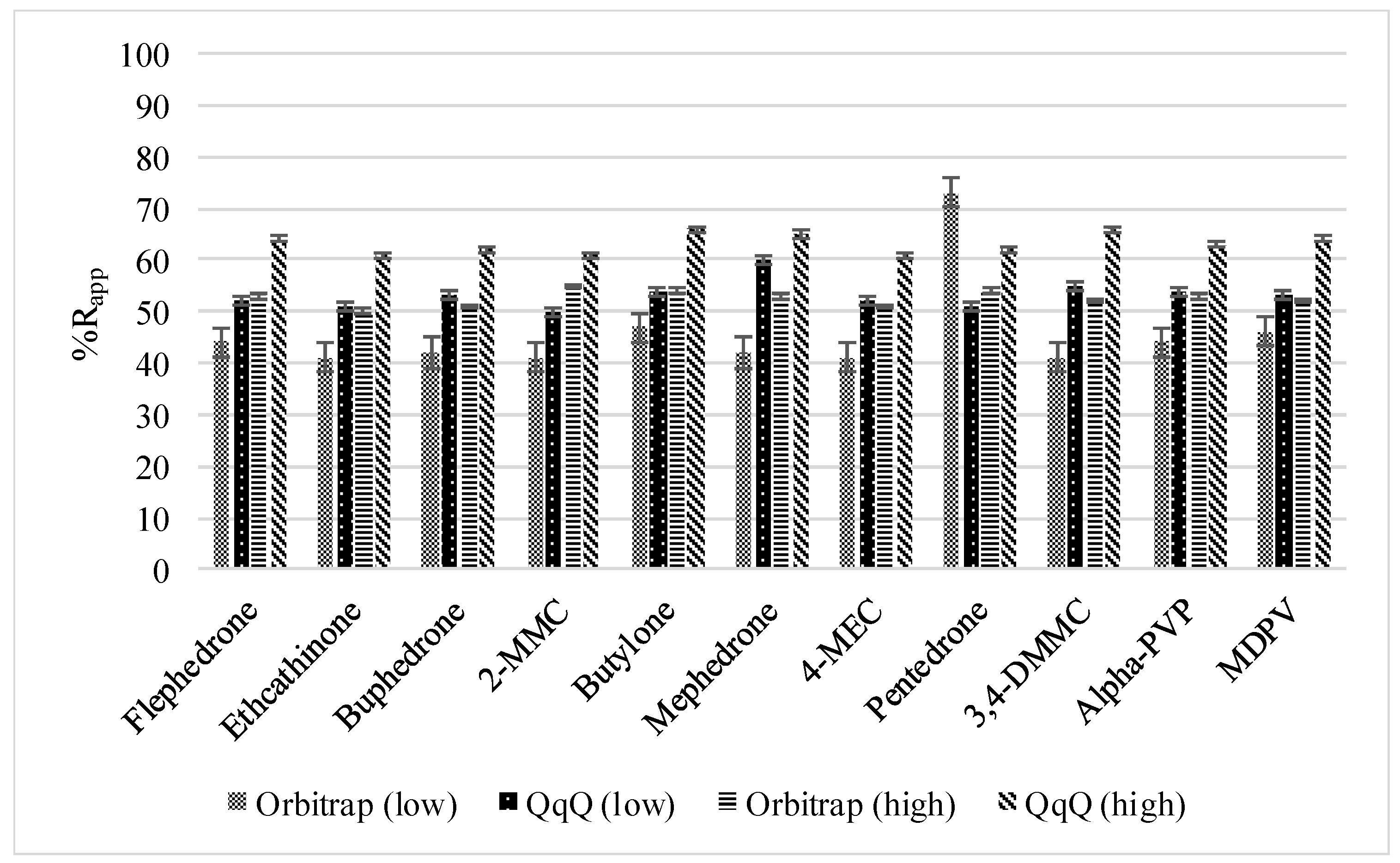
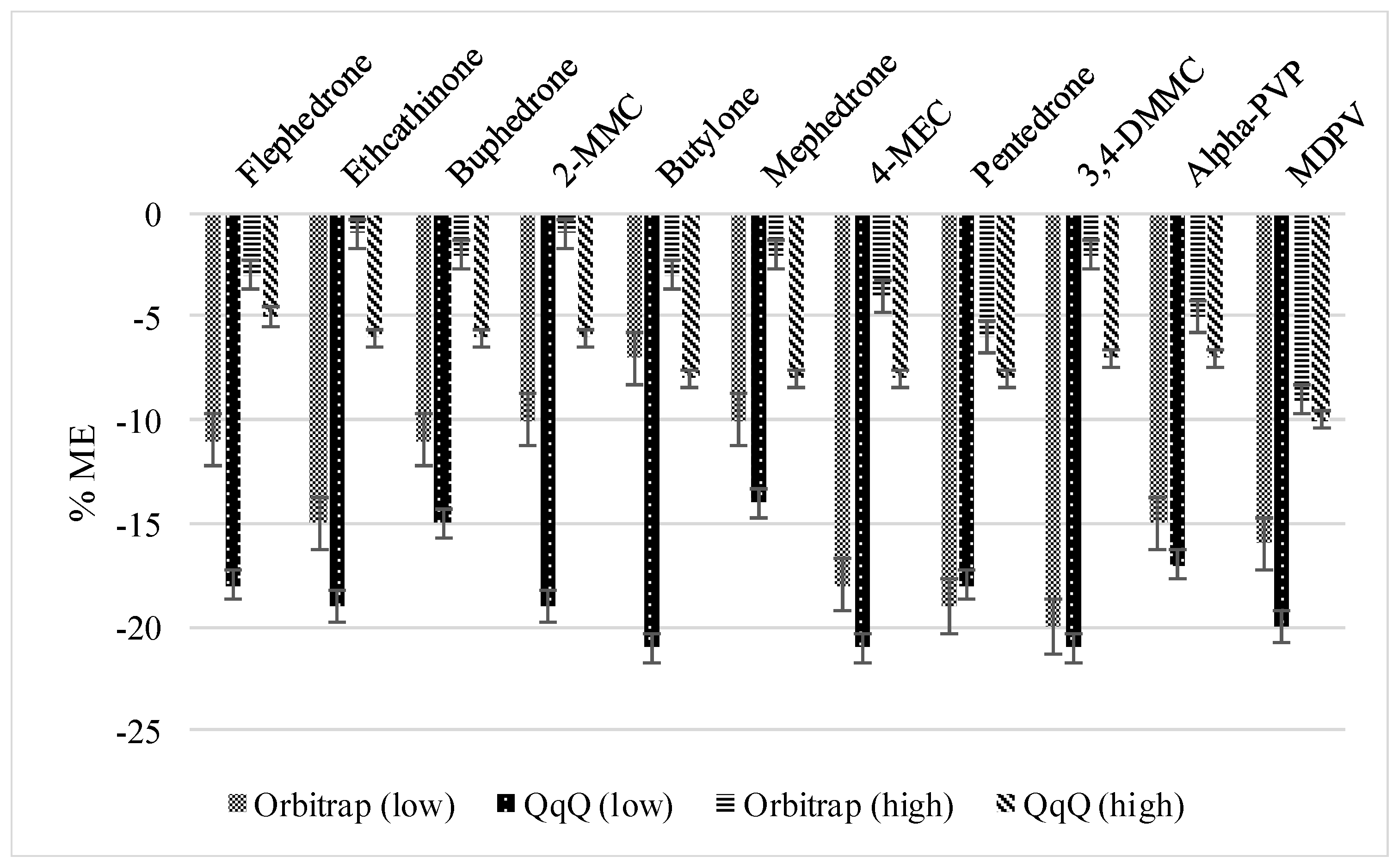
3.2.1. Method Recoveries and Matrix Effects
3.2.2. Linearity, MDLs, and MQLs
3.2.3. Accuracy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- United Nations Office on Drugs and Crime. Global Overview of Drug Demand and Supply. World Drug Report 2019. 2019. Available online: https://www.unodc.org/wdr2018/prelaunch/WDR18_Booklet_2_GLOBAL.pdf (accessed on 17 May 2020).
- United Nations Office on Drugs and Crime. World Drug Report 2019. Stimulants; UNITED NATIONS: 2019. Available online: https://www.unodc.org/wdr2018/prelaunch/WDR18_Booklet_3_DRUG_MARKETS.pdf (accessed on 17 May 2020).
- Valente, M.J.; Guedes De Pinho, P.; De Lourdes Bastos, M.; Carvalho, F.; Carvalho, M. Khat and synthetic cathinones: A review. Arch. Toxicol. 2014, 88, 15–45. [Google Scholar] [CrossRef]
- United Nations Office on Drugs and Crime. Drugs of Abuse 2017 Edition: A DEA Resource Guide. Forensic Toxicol. 2017, 37, 2352–2362. [Google Scholar] [CrossRef] [Green Version]
- European Monitoring Centre for Drugs and Drug Addiction EU Drug Markets Report 2019. 2019. Available online: http://www.emcdda.europa.eu/system/files/publications/12078/20192630_TD0319332ENN_PDF.pdf (accessed on 17 May 2020).
- Strickland, E.C.; Cummings, O.T.; Mellinger, A.L.; McIntire, G.L. Development and validation of a novel all-inclusive LC–MS-MS designer drug method. J. Anal. Toxicol. 2018, 43, 161–169. [Google Scholar] [CrossRef] [PubMed]
- de Castro, A.; Lendoiro, E.; Fernández-Vega, H.; Steinmeyer, S.; López-Rivadulla, M.; Cruz, A. Liquid chromatography tandem mass spectrometry determination of selected synthetic cathinones and two piperazines in oral fluid. Cross reactivity study with an on-site immunoassay device. J. Chromatogr. A 2014, 1374, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Strano-Rossi, S.; Anzillotti, L.; Castrignanò, E.; Felli, M.; Serpelloni, G.; Mollica, R.; Chiarotti, M. UHPLC-ESI-MS/MS method for direct analysis of drugs of abuse in oral fluid for DUID assessment. Anal. Bioanal. Chem. 2011, 401, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Strano-Rossi, S.; Anzillotti, L.; Castrignanò, E.; Romolo, F.S.; Chiarotti, M. Ultra high performance liquid chromatography-electrospray ionization-tandem mass spectrometry screening method for direct analysis of designer drugs, “spice” and stimulants in oral fluid. J. Chromatogr. A 2012, 1258, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Fernández, P.; Regenjo, M.; Ares, A.; Fernández, A.M.; Lorenzo, R.A.; Carro, A.M. Simultaneous determination of 20 drugs of abuse in oral fluid using ultrasound-assisted dispersive liquid–liquid microextraction. Anal. Bioanal. Chem. 2019, 411, 193–203. [Google Scholar] [CrossRef]
- Rocchi, R.; Simeoni, M.C.; Montesano, C.; Vannutelli, G.; Curini, R.; Sergi, M.; Compagnone, D.; Simeoni, M.C.; Rocchi, R.; Curini, R.; et al. Analysis of new psychoactive substances in oral fluids by means of microextraction by packed sorbent followed by ultra-high-performance liquid chromatography-tandem mass spectrometry. Drug Test. Anal. 2018, 10, 865–873. [Google Scholar] [CrossRef]
- Sorribes-Soriano, A.; Esteve-Turrillas, F.A.; Armenta, S.; Amorós, P.; Herrero-Martínez, J.M. Amphetamine-type stimulants analysis in oral fluid based on molecularly imprinting extraction. Anal. Chim. Acta 2019, 1052, 73–83. [Google Scholar] [CrossRef]
- Concheiro, M.; Anizan, S.; Ellefsen, K.; Huestis, M.A. Simultaneous quantification of 28 synthetic cathinones and metabolites in urine by liquid chromatography-high resolution mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 9437–9448. [Google Scholar] [CrossRef]
- Concheiro, M.; Castaneto, M.; Kronstrand, R.; Huestis, M.A. Simultaneous determination of 40 novel psychoactive stimulants in urine by liquid chromatography-high resolution mass spectrometry and library matching. J. Chromatogr. A 2015, 1397, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glicksberg, L.; Bryand, K.; Kerrigan, S. Identification and quantification of synthetic cathinones in blood and urine using liquid chromatography-quadrupole/time of flight (LC-Q/TOF) mass spectrometry. J. Chromatogr. B 2016, 1035, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Ippisch, J.; Herrmann, C.; Guber, S.; Schultis, W. Analysis of new designer drugs and common drugs of abuse in urine by a combined targeted and untargeted LC-HR-QTOFMS approach. Anal. Bioanal. Chem. 2014, 406, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Caro, S.; Fontanals, N.; Borrull, F.; Aguilar, C.; Calull, M. Solid-phase extraction based on cation-exchange sorbents followed by liquid chromatography high-resolution mass spectrometry to determine synthetic cathinones in urine. Forensic Toxicol. 2020, 38, 185–194. [Google Scholar] [CrossRef]
- Odoardi, S.; Valentini, V.; De Giovanni, N.; Pascali, V.L.; Strano-Rossi, S. High-throughput screening for drugs of abuse and pharmaceutical drugs in hair by liquid-chromatography-high resolution mass spectrometry (LC-HRMS). Microchem. J. 2017, 133, 302–310. [Google Scholar] [CrossRef]
- Pascual-Caro, S.; Borrull, F.; Calull, M.; Aguilar, C. A fast analytical method for determining synthetic cathinones in oral fluid by liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2020. accepted. [Google Scholar]
- Mercolini, L.; Protti, M.; Catapano, M.C.; Rudge, J.; Sberna, A.E. LC-MS/MS and volumetric absorptive microsampling for quantitative bioanalysis of cathinone analogues in dried urine, plasma and oral fluid samples. J. Pharm. Biomed. Anal. 2016, 123, 186–194. [Google Scholar] [CrossRef]
- Substance Abuse and Mental Health Services Administration. Clinical Drug Testing in Primary Care; Lulu.com: Morrisville, NC, USA, 2012. [Google Scholar]
- Dinis-Oliveira, R.J.; Vieira, D.N.; Magalhães, T. Guidelines for Collection of Biological Samples for Clinical and Forensic Toxicological Analysis. Forensic Sci. Res. 2016, 1, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Hadland, S.E.; Levy, S. Objective testing: Urine and other drug tests. Child Adolesc. Psychiatr. Clin. N. Am. 2016, 25, 549–565. [Google Scholar] [CrossRef] [Green Version]
- Wille, S.M.R.; Baumgartner, M.R.; Di Fazio, V.; Samyn, N.; Kraemer, T. Trends in drug testing in oral fluid and hair as alternative matrices. Bioanalysis 2014, 6, 2193–2209. [Google Scholar] [CrossRef]
- Øiestad, E.L.; Øiestad, Å.M.L.; Gjelstad, A.; Karinen, R. Oral fluid drug analysis in the age of new psychoactive substances. Bioanalysis 2016, 8, 691–710. [Google Scholar] [CrossRef]
- Institóris, L.; Árok, Z.; Seprenyi, K.; Varga, T.; Sára-Klausz, G.; Keller, É.; Tóth, R.A.; Sala, L.; Kereszty, É.; Róna, K. Frequency and structure of stimulant designer drug consumption among suspected drug users in Budapest and South-East Hungary in 2012–2013. Forensic Sci. Int. 2015, 248, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Herrero, P.; Cortés-Francisco, N.; Borrull, F.; Caixach, J.; Pocurull, E.; Marcé, R.M. Comparison of triple quadrupole mass spectrometry and Orbitrap high-resolution mass spectrometry in ultrahigh performance liquid chromatography for the determination of veterinary drugs in sewage: Benefits and drawbacks. J. Mass Spectrom. 2014, 49, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Henry, H.; Sobhi, H.R.; Scheibner, O.; Bromirski, M.; Nimkar, S.B.; Rochat, B. Comparison between a high-resolution single-stage Orbitrap and a triple quadrupole mass spectrometer for quantitative analyses of drugs. Rapid Commun. Mass Spectrom. 2012, 26, 499–509. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Quantitative and confirmative performance of liquid chromatography coupled to high-resolution mass spectrometry compared to tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 979–992. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Quantification of anthelmintic drug residues in milk and muscle tissues by liquid chromatography coupled to Orbitrap and liquid chromatography coupled to tandem mass spectrometry. Talanta 2011, 85, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Vanhaecke, L.; Van Meulebroek, L.; De Clercq, N.; Vanden Bussche, J. High resolution orbitrap mass spectrometry in comparison with tandem mass spectrometry for confirmation of anabolic steroids in meat. Anal. Chim. Acta 2013, 767, 118–127. [Google Scholar] [CrossRef]
- Bruce, S.J.; Rochat, B.; Béguin, A.; Pesse, B.; Guessous, I.; Boulat, O.; Henry, H. Analysis and quantification of vitamin D metabolites in serum by ultra-performance liquid chromatography coupled to tandem mass spectrometry and high-resolution mass spectrometry—A method comparison and validation. Rapid Commun. Mass Spectrom. 2013, 27, 200–206. [Google Scholar] [CrossRef]
- De Baere, S.; Osselaere, A.; Devreese, M.; Vanhaecke, L.; De Backer, P.; Croubels, S. Development of a liquid-chromatography tandem mass spectrometry and ultra-high-performance liquid chromatography high-resolution mass spectrometry method for the quantitative determination of zearalenone and its major metabolites in chicken and pig plasma. Anal. Chim. Acta 2012, 756, 37–48. [Google Scholar] [CrossRef]
- Gómez-Canela, C.; Cortés-Francisco, N.; Ventura, F.; Caixach, J.; Lacorte, S. Liquid chromatography coupled to tandem mass spectrometry and high resolution mass spectrometry as analytical tools to characterize multi-class cytostatic compounds. J. Chromatogr. A 2013, 1276, 78–94. [Google Scholar] [CrossRef]
- Viaene, J.; Lanckmans, K.; Dejaegher, B.; Mangelings, D.; Vander Heyden, Y. Comparison of a triple-quadrupole and a quadrupole time-of-flight mass analyzer to quantify 16 opioids in human plasma. J. Pharm. Biomed. Anal. 2016, 127, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Jemal, M.; Ramagiri, S.; Xia, Y.Q.; Humpreys, W.G.; Olah, T.; Korfmacher, W.A. It is time for a paradigm shift in drug discovery bioanalysis: From SRM to HRMS. J. Mass Spectrom. 2011, 46, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Sancho, J.V.; Ibáñez, M. Chapter 10. Multiresidue Analysis of Pesticides: LC–MS/MS versus LC–HRMS. In Fast Liquid Chromatography-Mass Spectrometry Methods in Food and Environmental Analysis; World Scientific: Singapore, 2015; pp. 381–419. [Google Scholar] [CrossRef]
- United Nations Office on Drugs and Crime. Laboratory and Scientific Section. Guidance for the Validation of Analytical Methodology and Calibration of Equipment Used for Testing of Illicit Drugs in Seized Materials and Biological Specimens: A Commitment to Quality and Continuous Improvement; United Nations Publications: New York, NY, USA, 2009; ISBN 9789211482430. [Google Scholar]
- Brcak, M.; Beck, O.; Bosch, T.; Carmichael, D.; Fucci, N.; George, C.; Piper, M.; Salomone, A.; Schielen, W.; Steinmeyer, S.; et al. European guidelines for workplace drug testing in oral fluid. Drug Test. Anal. 2018, 10, 402–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EC/96/23 COMMISSION DECISION of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Available online: https://eur-lex.europa.eu/legal-ontent/EN/TXT/PDF/?uri=CELEX:32002D0657&from=EN (accessed on 17 May 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protonated Molecule [M+H]+ | Fragment | |||||
|---|---|---|---|---|---|---|
| Analyte | Rt (min) | Formula | Accurate Mass m/z | Formula | Accurate Mass m/z | Ratio |
| Flephedrone | 6.17 | C10H13FNO | 182.09757 | C10H11FN | 164.08755 | 38 |
| C9H8FN | 149.06408 | 10 | ||||
| Ethcathinone | 6.29 | C11H16NO | 178.12264 | C11H14N | 160.11262 | 23 |
| C9H10N | 132.08132 | 12 | ||||
| Buphedrone | 7.66 | C11H16NO | 178.12264 | C11H14N | 160.11262 | 19 |
| C9H9N | 131.07350 | 6 | ||||
| 2-MMC | 8.97 | C11H16NO | 178.12264 | C11H14N | 160.11262 | 30 |
| C10H11N | 145.08915 | 16 | ||||
| Butylone | 9.05 | C12H16NO3 | 222.11247 | C11H12NO | 174.09189 | 14 |
| C12H14NO2 | 204.10245 | 17 | ||||
| Mephedrone | 9.38 | C11H16NO | 178.12264 | C11H14N | 160.11262 | 34 |
| C10H11N | 145.08915 | 14 | ||||
| 4-MEC | 10.58 | C12H18NO | 192.13829 | C12H16N | 174.12827 | 30 |
| C10H11N | 145.08915 | 9 | ||||
| Pentedrone | 10.98 | C12H18NO | 192.13829 | C12H16N | 174.12827 | 16 |
| C9H10N | 132.08132 | 10 | ||||
| 3,4-DMMC | 11.66 | C12H18NO | 192.13829 | C12H16N | 174.12827 | 37 |
| C11H13N | 159.10480 | 12 | ||||
| Alpha-PVP | 12.21 | C15H22NO | 232.16959 | C7H7 | 91.05478 | 15 |
| C7H5O | 105.03404 | 5 | ||||
| MDPV | 12.58 | C16H22NO3 | 276.15942 | C8H16N | 126.12827 | 8 |
| C8H7O2 | 135.04461 | 9 | ||||
| Analyte | Rt (min) | Fragmentor (V) | Precursor Ion | Product Ion | CE (eV) | Ion Ratio |
|---|---|---|---|---|---|---|
| Flephedrone | 6.53 | 100 | 182.1 | 164 | 15 | |
| 149 | 15 | 70 | ||||
| 123 | 15 | 21 | ||||
| Ethcathinone | 6.62 | 100 | 178.1 | 160 | 12 | |
| 132 | 14 | 93 | ||||
| 105 | 18 | 39 | ||||
| Buphedrone | 8.29 | 100 | 178.1 | 160 | 10 | |
| 131 | 15 | 55 | ||||
| 132 | 15 | 52 | ||||
| 2-MMC | 9.98 | 100 | 178.1 | 160 | 8 | |
| 145 | 15 | 71 | ||||
| 119 | 15 | 11 | ||||
| Butylone | 10.26 | 100 | 222.1 | 174 | 14 | |
| 204 | 10 | 101 | ||||
| 191 | 10 | 23 | ||||
| Mephedrone | 10.48 | 100 | 178.1 | 160 | 8 | |
| 145 | 15 | 61 | ||||
| 119 | 15 | 12 | ||||
| 4-MEC | 11.81 | 100 | 192.1 | 174 | 10 | |
| 146 | 16 | 21 | ||||
| 145 | 18 | 24 | ||||
| Pentedrone | 12.25 | 100 | 192.1 | 174 | 10 | |
| 132 | 16 | 49 | ||||
| 91 | 18 | 24 | ||||
| 3,4-DMMC | 13.02 | 125 | 192.1 | 174 | 10 | |
| 159 | 15 | 57 | ||||
| 133 | 15 | 7 | ||||
| Alpha-PVP | 13.50 | 125 | 232.2 | 91 | 22 | |
| 105 | 25 | 42 | ||||
| 126 | 25 | 41 | ||||
| MDPV | 13.96 | 125 | 276.2 | 126 | 25 | |
| 135 | 25 | 67 | ||||
| 175 | 20 | 52 |
| MDL Orbitrap | MDL QqQ | MQL Orbitrap | MQL QqQ | Linear Range Orbitrap | Linear Range QqQ | |
|---|---|---|---|---|---|---|
| Flephedrone | 0.080 | 0.025 | 0.200 | 0.050 | 0.200–4⋯4–200 | 0.050–4⋯4–200 |
| Ethcathinone | 0.100 | 0.030 | 0.050 | 0.050 | 0.050–4⋯4–200 | 0.050–4⋯4–200 |
| Buphedrone | 0.100 | 0.035 | 0.200 | 0.050 | 0.200–4⋯4–200 | 0.050–4⋯4–200 |
| 2-MMC | 0.100 | 0.025 | 0.050 | 0.050 | 0.050–4⋯4–200 | 0.050–4⋯4–200 |
| Butylone | 0.120 | 0.030 | 0.050 | 0.050 | 0.050–4⋯4–200 | 0.050–4⋯4–200 |
| Mephedrone | 0.080 | 0.005 | 0.050 | 0.030 | 0.050–4⋯4–200 | 0.030–4⋯4–200 |
| 4-MEC | 0.040 | 0.010 | 0.200 | 0.030 | 0.200–4⋯4–200 | 0.030–4⋯4–200 |
| Pentedrone | 0.040 | 0.025 | 0.050 | 0.030 | 0.050–4⋯4–200 | 0.030–4⋯4–200 |
| 3,4-DMMC | 0.040 | 0.010 | 0.050 | 0.030 | 0.050–4⋯4–200 | 0.030–4⋯4–200 |
| Alpha-PVP | 0.160 | 0.005 | 0.050 | 0.020 | 0.050–4⋯4–200 | 0.020–4⋯4–200 |
| MDPV | 0.060 | 0.005 | 0.050 | 0.030 | 0.050–4⋯4–200 | 0.030–4⋯4–200 |
| MDL Orbitrap | MDL QqQ | MQL Orbitrap | MQL QqQ | Linear Range Orbitrap | Linear Range QqQ | |
|---|---|---|---|---|---|---|
| Flephedrone | 0.020 | 0.010 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075–4⋯4–75 |
| Ethcathinone | 0.035 | 0.030 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075–4⋯4–75 |
| Buphedrone | 0.025 | 0.030 | 0.100 | 0.075 | 0.100–4⋯4–75 | 0.075–4⋯4–75 |
| 2-MMC | 0.015 | 0.015 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075–4⋯4–75 |
| Butylone | 0.020 | 0.010 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075–4⋯4–75 |
| Mephedrone | 0.010 | 0.010 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075–4⋯4–75 |
| 4-MEC | 0.025 | 0.020 | 0.100 | 0.075 | 0.100–4⋯4–75 | 0.075–4⋯4–75 |
| Pentedrone | 0.020 | 0.020 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075–4⋯4–75 |
| 3,4-DMMC | 0.015 | 0.010 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075–4⋯4–75 |
| Alpha-PVP | 0.010 | 0.005 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075 –4⋯4–75 |
| MDPV | 0.015 | 0.003 | 0.075 | 0.075 | 0.075–4⋯4–75 | 0.075–4⋯4–75 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascual-Caro, S.; Borrull, F.; Aguilar, C.; Calull, M. Determination of Synthetic Cathinones in Urine and Oral Fluid by Liquid Chromatography High-Resolution Mass Spectrometry and Low-Resolution Mass Spectrometry: A Method Comparison. Separations 2020, 7, 53. https://doi.org/10.3390/separations7040053
Pascual-Caro S, Borrull F, Aguilar C, Calull M. Determination of Synthetic Cathinones in Urine and Oral Fluid by Liquid Chromatography High-Resolution Mass Spectrometry and Low-Resolution Mass Spectrometry: A Method Comparison. Separations. 2020; 7(4):53. https://doi.org/10.3390/separations7040053
Chicago/Turabian StylePascual-Caro, Sergi, Francesc Borrull, Carme Aguilar, and Marta Calull. 2020. "Determination of Synthetic Cathinones in Urine and Oral Fluid by Liquid Chromatography High-Resolution Mass Spectrometry and Low-Resolution Mass Spectrometry: A Method Comparison" Separations 7, no. 4: 53. https://doi.org/10.3390/separations7040053
APA StylePascual-Caro, S., Borrull, F., Aguilar, C., & Calull, M. (2020). Determination of Synthetic Cathinones in Urine and Oral Fluid by Liquid Chromatography High-Resolution Mass Spectrometry and Low-Resolution Mass Spectrometry: A Method Comparison. Separations, 7(4), 53. https://doi.org/10.3390/separations7040053