Analysis of Monoclonal Antibodies by Capillary Electrophoresis: Sample Preparation, Separation, and Detection


Abstract
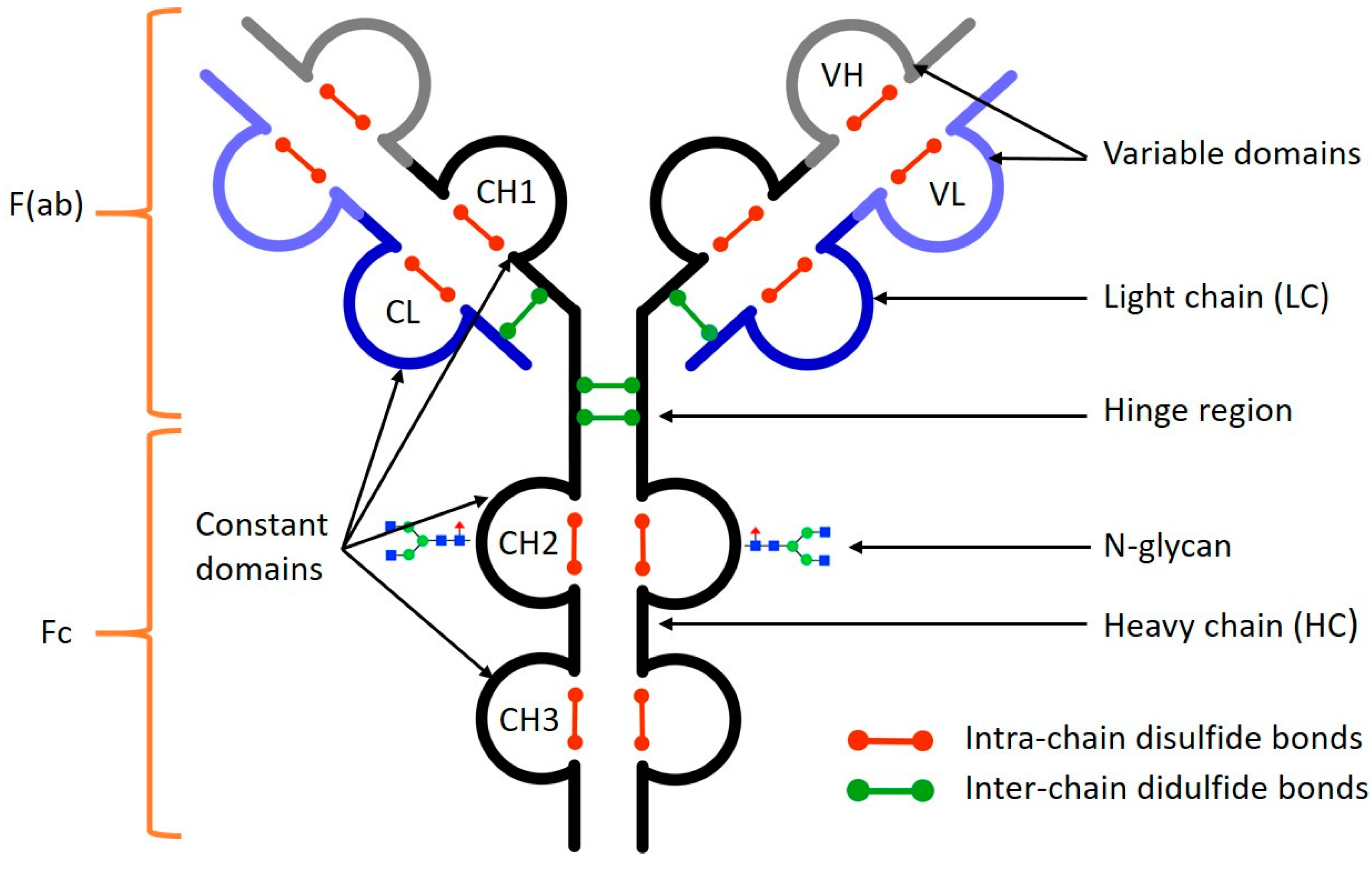
:1. Introduction
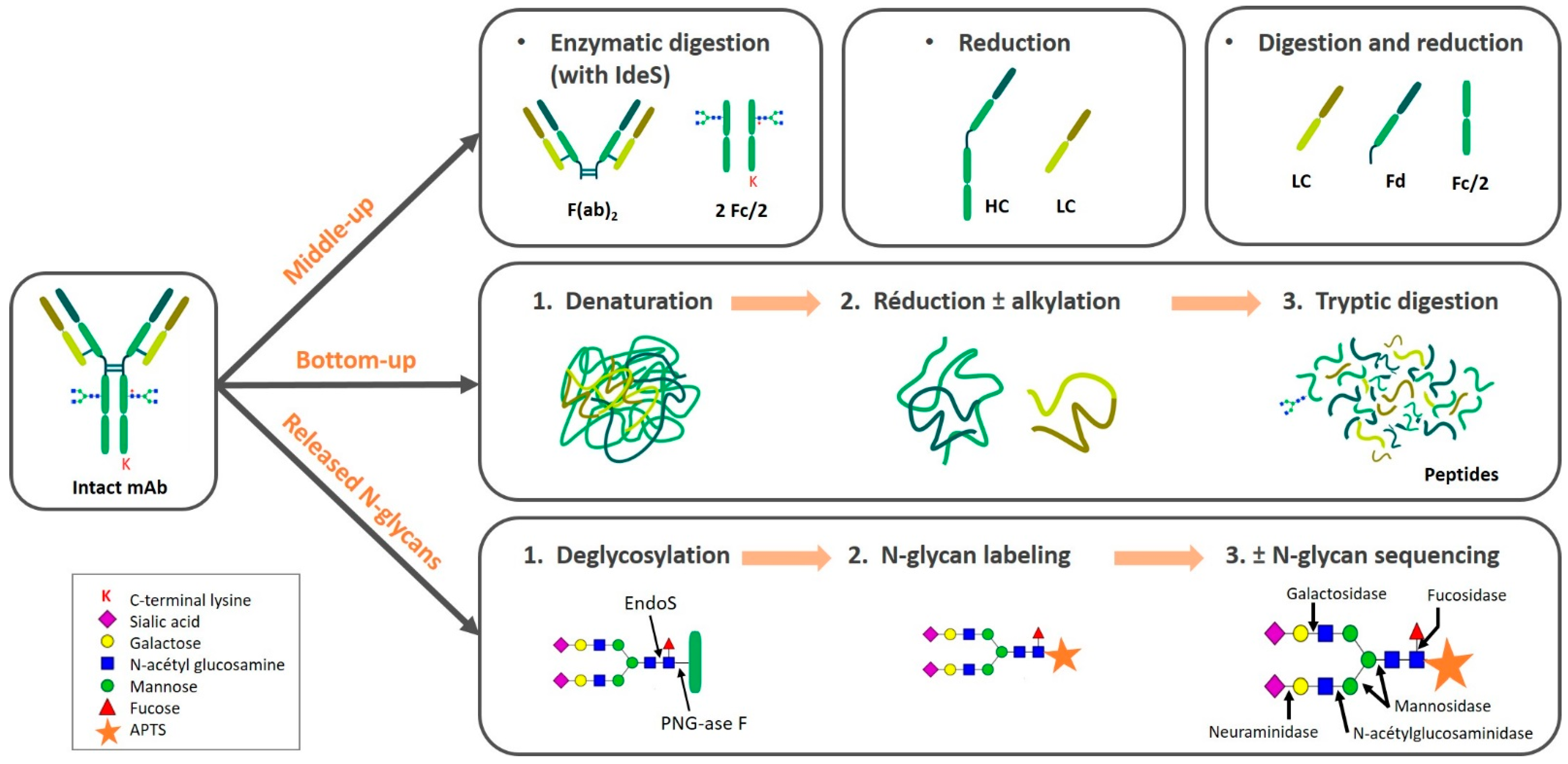
2. Sample Preparation of Monoclonal Antibodies
2.1. Analytical Considerations
2.2. Sample Preparation Approaches
2.2.1. Intact and Top-Down
2.2.2. Middle-Up/Down
Enzymatic Digestion
Reduction
Multiple Reactions
2.2.3. Bottom-Up
Denaturation
- -
- Urea destabilizes mAbs by the formation of hydrogen bonds [35]. However, it decomposes into isocyanic acid with time and heat, causing mAbs carbamylation that induces mass artifacts of 43 Da [36]. Carbamylation depends on temperature, incubation time, and pH. It can be minimized by using a buffer containing ammonium ions [37];
- -
- -
- RapiGest® (sodium 3-((2-methyl-2-undecyl-1,3-dioxolan-4-yl)methoxyl)-1-propane sulfonate) is an efficient surfactant compatible with different enzymes. It is also compatible with MS analysis due to the fact of its acid-labile character [40]. It causes protein unfolding and is cleaved by adding formic acid at the end of digestion. Consequently, it does not interfere with electrophoretic separation and MS detection.
Reduction and Alkylation
Digestion
2.2.4. Released N-glycans
Deglycosylation
N-glycan Labeling
N-glycan Sequencing
3. Capillary Electrophoresis Analyses
3.1. Capillary Zone Electrophoresis (CZE)
3.1.1. Technical Considerations
- Capillary coating
- MS-based detection modes
3.1.2. CZE Analyses
Intact and Middle-Up Approaches
- Analyses under non-compatible conditions with MS detectionCapillary zone electrophoresis-UV is frequently used to evaluate charge heterogeneity of several mAbs at the intact and middle-up levels in routine analyses [73,88,89]. An intercompany study has demonstrated its high performance for analyses of a broad range of mAb pI and its suitability for good manufacturing practice (GMP) environment [88]. A comparative study revealed its higher performance on the separation of charge variants over cIEF [90]. He et al. showed that the charge differences between variants are pH dependent and their separation occurs at pH closer to the pI of mAbs. Furthermore, mAbs have a limited solubility at pH around their pI and tend to precipitate [91]. Consequently, the pH of the BGE and the sample buffer should be chosen carefully.6-Aminocaproic acid-based BGE is a standard BGE for mAbs analyses with CZE–UV due to the fact of its zwitterionic properties and low conductivity [92,93]. It is often used at high ionic strength to lower EOF and enhance separation resolution [73]. 6-Aminocaproic acid/citric acid at lower ionic strength provided a high resolution of separation and peak efficiencies for analyses of infliximab subunits after IdeS digestion [94]. Additives can be added in the sample buffer or BGE to enhance peak shape or resolution. Urea was added to BGE to partially separate disulfide isomers of an IgG2 mAb [95]. Several studies reported the optimization of BGE composition. Suba et al. used a “two-phase-four-step” approach for rapid optimization of BGE (6-aminocaproic acid, TETA, and HPMC) for analyses of intact and papain digested mAb samples [96]. Moritz et al. applied the design of experiments approach (DoE) for the optimization of analysis conditions. The impact of several parameters of BGE, including TETA concentration, pH value, polymer additive (HPC versus HPMC), and other additives (butanolamine and acetonitrile) were investigated [97].Despite its high performance and cost-effectiveness, CZE–UV does not allow the identification of separated peaks. The conditions above cannot be used in CE–ESI–MS as 6-aminocaproic acid-based BGEs are not volatiles. A heart-cut CZE–CZE–MS system was developed to perform separation and quantitation of charge variants by 6-aminocaproic acid-based CZE–UV by the first dimension and acetic acid-based CZE–MS to establish their identities and modifications [92]. Asymmetric conditions of BGE have been developed to characterize digested cetuximab with IdeS by off-line CZE–UV coupled to MALDI-MS via a fraction collection platform [98,99]. Inlet BGE was based on 6-aminocaproic acid/acetic acid; outlet BGE was based on ammonium acetate to allow good crystallization of collected fractions in the matrix 2,5-dihydroxybenzoic acid in 0.1% trifluoroacetic acid/acetonitrile (TFA/ACN; 30/70, v/v) deposited on the MALDI plate. The developed conditions allowed the separation of F(ab)2 glycoforms with NGNA residue [24,99].
- Analyses under MS-compatible conditionsThe CZE–ESI–MS system requires the use of volatile solutions and is incompatible with the presence of salts that would cause adduct formation and ion suppression. As a consequence, the choice of the sample matrix and BGE is pretty limited for CZE–ESI–MS. Acetic acid, formic acid, and ammonium acetate are typical for the analyses of mAbs in CZE–ESI–MS.
- ○
- Denaturing conditionsDenaturing conditions of sample buffer and BGE cause the mAbs unfolding and dissociate non-covalent interactions. Consequently, ESI efficiency is enhanced (an increase of charge state) and proper investigation of microheterogeneities is done.Samples are sometimes prepared in denaturing solutions that consist in aqueous solutions of acid and organic solvent such as acetonitrile, methanol, and isopropanol to reduce sample zone conductivity and enhance the resolution of separation. The presence of reduced mAbs in an aqueous solution of 35% acetic acid/50% acetonitrile (v/v) favored sample stacking and allowed the injection of 7% of the capillary volume [100]. A 0.2% acetic acid/10% isopropanol (v/v) aqueous solution was used as a sample buffer and BGE for analyses of different intact mAbs and an ADC in a microfluidic system coupled to MS. The results allowed the characterization of partially separated charge variants as clipping of C-terminal lysine and their related glycoforms. In addition to these pieces of information, the drug-to-antibody ratio was determined after full CZE separation of the ADC charge variants [93,101]. In another study, an aqueous solution of 15% acetic acid/15% acetonitrile (v/v) was used as a sample buffer for high resolution of separation of subunits of reduced, digested, digested-reduced mAb as well as their characterization with sheath-liquid CZE–MS and neutral coating [102].Acidic BGE (pH < 3) is often used to separate variants or subunits of mAbs and enhance ESI efficiency. Acetic acid is used instead of formic acid for CZE separations due to its very low conductivity that avoid joule heating [103]. Additives such as organic solvents are sometimes used to enhance variants solubility and decrease hydrophobic interactions between them [97]. Acetic acid BGE was used to perform sheathless CE–MS with a neutral coating to perform separation and assignment of intact nanobodies and their deamidated and truncated forms at the C-terminal tag. Moreover, isomeric deamidated products were efficiently separated. The heterogeneity of intact and IdeS digested mAbs (trastuzumab, infliximab, and ustekinumab) was also investigated. The F(ab)2 and Fc/2 fragments were highly resolved and partial separation of Fc/2 charge variants was obtained [75]. Georgetti et al. optimized the composition of sample buffer and % of acetic acid BGE. Intact or digested mAbs were prepared in an aqueous solution of 1% formic acid/30% methanol (v/v) after desalting and 3% acetic acid was used as BGE. These conditions allowed the partial separation of charge variants, di-glycosylated, and mono-glycosylated forms of different intact mAbs. In addition, complete or partial separation and characterization were achieved for IdeS digestion products, IdeS digestion followed by reduction products as well as their variants with positively charged coating [104,105]. Belov et al. performed high resolution separation of charge variants, deamidated forms, sialylated, and non-sialylated glycoforms with MS/MS confirmation at the middle-down level. Even bi-, mono- and non-glycosylated forms of a mAb were separated at the intact level using 0.2% formic acid/10% isopropanol (v/v) BGE and M7C4I coating [76]. Nevertheless, unwanted modifications of microvariations can be induced by the exposure of mAbs to these conditions, such as loss of sialic acid of N-glycans, which is considered as a critical quality attribute [106,107].
- ○
- Non-denaturing conditionsAmmonium acetate is the core sample buffer and BGE for native or near-native CZE-ESI-MS characterization (pH 5.0–7.0). It preserves non-covalent interactions and conformational heterogeneity of mAbs due to the fact of its volatility. Francois et al. characterized the Fc/2 dimers formed by non-covalent interactions by CE–MS [24]. They used ammonium acetate as a sample buffer for sheathless CE–MS infusion of collected fractions after CZE separation. Said et al. performed a sheathless CE–MS infusion to estimate the drug load distribution and the drug-to-antibody ratio (DAR) of intact ADC (bretuximab-vedotin) as well as its subunits after IdeS digestion by sheathless CE–MS [108]. Dadouch et al. used ammonium acetate both as BGE and proteolysis buffer for in-line middle-up analyses of infliximab because of its compatibility with IdeS digestion. They implemented the in-line methodology with TDLFP mixing and simplified procedure using plug–plug mode for reactants injection and temperature control. The methodology provided complete in-line digestion and separation, a significant decrease in reactant consumption and digestion time, and higher peak efficiency comparing with the off-line assay. The digestion products and Fc/2 dimers were identified by sheathless CE–MS [94]. Carillo et al. achieved partial separation of charge variants of trastuzumab and bevacizumab by µCE–ESI–MS with ammonium acetate/DMSO BGE analyses. Charge variants profiles were determined and related glycoforms, particularly sialylated N-glycans, were identified and relatively quantitated with high accuracy and sensitivity [109]. Ammonium acetate has also been employed for the assessment of mAbs stability and aggregate formation. Belov et al. characterized non-covalent dimeric aggregates, their glycosylation profile and the glycan pairing of intact trastuzumab [110]. Minh et al. studied the unfolding and aggregation of stressed infliximab. Folded and unfolded infliximab were partially separated. They investigated infliximab dimers formation and attributed its formation to the interaction between unfolded infliximab molecules via F(ab) regions [111]. Characterization of mAbs by CE–ESI–MS under native conditions is still challenging due to the spray generation with pressure in the case of neutral coating or electroosmotic flow in the case of positively charged coating that compromises the separation efficiency.Intact and middle-up approaches can be followed by MS/MS analyses referred to top-down and middle-down to confirm results. These approaches allow mAbs sequencing by direct fragmentation of intact mAbs or mAbs subunits in ion source decay or higher collision energy dissociation cell of mass spectrometers [76,99]. As a consequence, they are powerful alternative approaches to the bottom-up approach to avoid sample preparation artifacts. However, it is still challenging to get complete sequence coverage due to the following technical issues: (i) the decrease of MS sensitivity with the increase of the molecular weight of analytes that affects the fragmentation efficiency and (ii) sophisticated mass spectrometers with high resolution are required [112].
Bottom-Up
Released N-glycans
Related Products Impurities
3.2. Capillary Gel Electrophoresis (CGE)
3.2.1. Non-Reduced CGE-SDS
3.2.2. Reduced CGE-SDS
3.2.3. Coupling with MS
3.2.4. CGE Analyses of Released N-glycans
3.3. Capillary Isoelectric Focusing (cIEF)
3.4. Micellar Electrokinetic Capillary Chromatography (MEKC)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bouzid, K.; Bedairia, N.; Marty, M. Anticorps monoclonaux thérapeutiques en cancérologie. Pathol. Biol. 2012, 60, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Tsumoto, K.; Isozaki, Y.; Yagami, H.; Tomita, M. Future perspectives of therapeutic monoclonal antibodies. Immunotherapy. 2019, 11, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.D.; Deng, R.; Boswell, C.A.; Zhang, Y.; Li, J.; Fielder, P.; Joshi, A.; Kenkare-Mitra, S. Monoclonal Antibodies: From Structure to Therapeutic Application. In Pharmaceutical Biotechnology; Springer: New York, NY, USA, 2013; pp. 143–178. [Google Scholar]
- Todoroki, K.; Yamada, T.; Mizuno, H.; Toyo’oka, T. Current Mass Spectrometric Tools for the Bioanalyses of Therapeutic Monoclonal Antibodies and Antibody-Drug Conjugates. Anal. Sci. 2018, 34, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wang, D.; Mason, B.; Rossomando, T.; Li, N.; Liu, D.; Cheung, J.K.; Xu, W.; Raghava, S.; Katiyar, A.; et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs 2019, 11, 239–264. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, D.; Jabs, W.; Resemann, A.; Evers, W.; Evans, C.; Main, L.; Baessmann, C.; Wagner-Rousset, E.; Suckau, D.; Beck, A. Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques. MAbs 2013, 5, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Cymer, F.; Beck, H.; Rohde, A.; Reusch, D. Therapeutic monoclonal antibody N-glycosylation—Structure, function and therapeutic potential. Biologicals 2018, 52, 1–11. [Google Scholar] [CrossRef]
- Beck, A.; Liu, H. Macro- and Micro-Heterogeneity of Natural and Recombinant IgG Antibodies. Antibodies 2019, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Torkashvand, F.; Vaziri, B. Main quality attributes of monoclonal antibodies and effect of cell culture components. Iran. Biomed. J. 2017, 21, 131–141. [Google Scholar] [CrossRef]
- Geigert, J.; Geigert, J. Quality Attributes of a Biopharmaceutical. In The Challenge of CMC Regulatory Compliance for Biopharmaceuticals; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; pp. 311–329. [Google Scholar]
- Parr, M.K.; Montacir, O.; Montacir, H. Physicochemical characterization of biopharmaceuticals. J. Pharm. Biomed. Anal. 2016, 130, 366–389. [Google Scholar] [CrossRef]
- Fekete, S.; Gassner, A.L.; Rudaz, S.; Schappler, J.; Guillarme, D. Analytical strategies for the characterization of therapeutic monoclonal antibodies. TrAC Trends Anal. Chem. 2013, 42, 74–83. [Google Scholar] [CrossRef]
- Gahoual, R.; Beck, A.; Leize-Wagner, E.; François, Y.-N. Cutting-edge capillary electrophoresis characterization of monoclonal antibodies and related products. J. Chromatogr. B 2016, 1032, 61–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, A.; Giorgetti, J.; Gahoual, R.; Beck, A.; Leize-Wagner, E.; François, Y.N. Insights from capillary electrophoresis approaches for characterization of monoclonal antibodies and antibody drug conjugates in the period 2016–2018. J. Chromatogr. B 2019, 1122–1123, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebbi, V.; Chattopadhyay, S.; Rathore, A.S. High performance liquid chromatography (HPLC) based direct and simultaneous estimation of excipients in biopharmaceutical products. J. Chromatogr. B 2019, 1117, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Falconer, R.J. Advances in liquid formulations of parenteral therapeutic proteins. Biotechnol. Adv. 2019, 37, 107412–107421. [Google Scholar] [CrossRef]
- Le Basle, Y.; Chennell, P.; Tokhadze, N.; Astier, A.; Sautou, V. Physicochemical Stability of Monoclonal Antibodies: A Review. J. Pharm. Sci. 2019, 109, 169–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Authelin, J.-R.; Rodrigues, M.A.; Tchessalov, S.; Singh, S.; McCoy, T.; Wang, S.; Shalaev, E. Freezing of biologicals revisited: Scale, stability, excipients, and degradation stresses. J. Pharm. Sci. 2020, 109, 44–61. [Google Scholar] [CrossRef]
- Bobaly, B.; D’Atri, V.; Goyon, A.; Colas, O.; Beck, A.; Fekete, S.; Guillarme, D. Protocols for the analytical characterization of therapeutic monoclonal antibodies. II—Enzymatic and chemical sample preparation. J. Chromatogr. B 2017, 1060, 325–335. [Google Scholar] [CrossRef]
- Beckers, J.L.; Boč Ek, P. The preparation of background electrolytes in capillary zone electrophoresis: Golden rules and pitfalls. Electrophoresis. 2003, 24, 518–535. [Google Scholar] [CrossRef]
- Chen, Y.; Mori, M.; Pastusek, A.C.; Schug, K.A.; Dasgupta, P.K. On-Line Electrodialytic Salt Removal in Electrospray Ionization Mass Spectrometry of Proteins. Anal. Chem. 2011, 83, 1015–1021. [Google Scholar] [CrossRef]
- Shire, S.J.; Shahrokh, Z.; Liu, J. Challenges in the development of high protein concentration formulations. J. Pharm. Sci. 2004, 93, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- François, Y.-N.; Biacchi, M.; Said, N.; Renard, C.; Beck, A.; Gahoual, R.; Leize-Wagner, E. Characterization of cetuximab Fc/2 dimers by off-line CZE-MS. Anal. Chim. Acta 2016, 908, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Livingston, E.; Chen, X. High Throughput Profiling of Charge Heterogeneity in Antibodies by Microchip Electrophoresis. Anal. Chem. 2011, 83, 8184–8191. [Google Scholar] [CrossRef] [PubMed]
- Oscar, S.-S.; Brandon, T.; Sarah, D.; Monica, P.; Alex, S.; Ma, S. Optimization and Validation of a Quantitative Capillary Electrophoresis Sodium Dodecyl Sulfate Method for Quality Control and Stability Monitoring of Monoclonal Antibodies. Anal. Chem. 2006, 78, 6583–6594. [Google Scholar] [CrossRef]
- Goyon, A.; D’Atri, V.; Bobaly, B.; Wagner-Rousset, E.; Beck, A.; Fekete, S.; Guillarme, D. Protocols for the analytical characterization of therapeutic monoclonal antibodies. I—Non-denaturing chromatographic techniques. J. Chromatogr. B 2017, 1058, 73–84. [Google Scholar] [CrossRef]
- Sjögren, J.; Andersson, L.; Mejàre, M.; Olsson, F. Generating and purifying Fab fragments from human and mouse IgG using the bacterial enzymes IdeS, SpeB and Kgp. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1535, pp. 319–329. [Google Scholar]
- Sjögren, J.; Olsson, F.; Beck, A. Rapid and improved characterization of therapeutic antibodies and antibody related products using IdeS digestion and subunit analysis. Analyst 2016, 141, 3114–3125. [Google Scholar] [CrossRef] [PubMed]
- Domkin, V.; Chabes, A. Phosphines are ribonucleotide reductase reductants that act via C-terminal cysteines similar to thioredoxins and glutaredoxins. Sci. Rep. 2015, 4, 5539. [Google Scholar] [CrossRef] [Green Version]
- Wenig, K.; Chatwell, L.; von Pawel-Rammingen, U.; Björck, L.; Huber, R.; Sondermann, P. Structure of the streptococcal endopeptidase IdeS, a cysteine proteinase with strict specificity for IgG. Proc. Natl. Acad. Sci. USA 2004, 101, 17371–17376. [Google Scholar] [CrossRef] [Green Version]
- Faid, V.; Leblanc, Y.; Bihoreau, N.; Chevreux, G. Middle-up analysis of monoclonal antibodies after combined IgdE and IdeS hinge proteolysis: Investigation of free sulfhydryls. J. Pharm. Biomed. Anal. 2018, 149, 541–546. [Google Scholar] [CrossRef]
- Thiede, B.; Höhenwarter, W.; Krah, A.; Mattow, J.; Schmid, M.; Schmidt, F.; Jungblut, P.R. Peptide mass fingerprinting. Methods 2005, 35, 237–247. [Google Scholar] [CrossRef]
- Gahoual, R.; Burr, A.; Busnel, J.-M.; Kuhn, L.; Hammann, P.; Beck, A.; François, Y.-N.; Leize-Wagner, E. Rapid and multi-level characterization of trastuzumab using sheathless capillary electrophoresis-tandem mass spectrometry. MAbs 2013, 5, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.K.; Rösgen, J.; Englander, S.W. Urea, but not guanidinium, destabilizes proteins by forming hydrogen bonds to the peptide group. Proc. Natl. Acad. Sci. USA 2009, 106, 2595–2600. [Google Scholar] [CrossRef] [Green Version]
- Dick, L.W.; Mahon, D.; Qiu, D.; Cheng, K.C. Peptide mapping of therapeutic monoclonal antibodies: Improvements for increased speed and fewer artifacts. J. Chromatogr. B 2009, 877, 230–236. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, J.Y.; Yang, W.; Zhang, H. Inhibition of protein carbamylation in urea solution using ammonium-containing buffers. Anal. Biochem. 2014, 446, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Pace, C.N.; Grimsley, G.R.; Scholtz, J.M. Denaturation of Proteins by Urea and Guanidine Hydrochloride. In Protein Science Encyclopedia; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Mouchahoir, T.; Schiel, J.E. Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Anal. Bioanal. Chem. 2018, 410, 2111–2126. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.-Q.; Gilar, M.; Lee, P.J.; Bouvier, E.S.P.; Gebler, J.C. Enzyme-Friendly, Mass Spectrometry-Compatible Surfactant for In-Solution Enzymatic Digestion of Proteins. Anal. Chem. 2003, 75, 6023–6028. [Google Scholar] [CrossRef]
- McNulty, J.; Krishnamoorthy, V.; Amoroso, D.; Moser, M. Tris(3-hydroxypropyl)phosphine (THPP): A mild, air-stable reagent for the rapid, reductive cleavage of small-molecule disulfides. Bioorganic Med. Chem. Lett. 2015, 25, 4114–4117. [Google Scholar] [CrossRef]
- Huang, H.Z.; Nichols, A.; Liu, D. Direct Identification and Quantification of Aspartyl Succinimide in an IgG2 mAb by RapiGest Assisted Digestion. Anal. Chem. 2009, 81, 1686–1692. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, M.T.; Patterson, L.C.; Yu, X.C.; Zhang, Y.T.; Burgess, B.L.; Chen, Y. Domain-specific free thiol variant characterization of an IgG1 by reversed-phase high-performance liquid chromatography mass spectrometry. Anal. Biochem. 2017, 519, 8–14. [Google Scholar] [CrossRef]
- Rebecchi, K.R.; Go, E.P.; Xu, L.; Woodin, C.L.; Mure, M.; Desaire, H. Protein Sequence Coverage and Post-Translational Modifications Analysis of Recombinant Glycoproteins: Application to the Characterization of Human Lysyl Oxidase. Anal. Chem. 2011, 83, 8484–8491. [Google Scholar] [CrossRef] [Green Version]
- Huhn, C.; Selman, M.H.J.; Ruhaak, L.R.; Deelder, A.M.; Wuhrer, M. IgG glycosylation analysis. Proteomics 2009, 9, 882–913. [Google Scholar] [CrossRef] [PubMed]
- Switzar, L.; Giera, M.; Niessen, W.M.A. Protein Digestion: An Overview of the Available Techniques and Recent Developments. J. Proteome Res. 2013, 12, 1067–1077. [Google Scholar] [CrossRef]
- Dada, O.O.; Rao, R.; Jones, N.; Jaya, N.; Salas-Solano, O. Comparison of SEC and CE-SDS methods for monitoring hinge fragmentation in IgG1 monoclonal antibodies. J. Pharm. Biomed. Anal. 2017, 145, 91–97. [Google Scholar] [CrossRef]
- Beck, A.; Diemer, H.; Ayoub, D.; Debaene, F.; Wagner-Rousset, E.; Carapito, C.; Van Dorsselaer, A.; Sanglier-Cianférani, S. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. TrAC Trends Anal. Chem. 2013, 48, 81–95. [Google Scholar] [CrossRef]
- Hansen, K.; Szarka, S.; Escoffier, E.; Berthet, A.; Venet, J.; Collet-Brose, J.; Hepburn, S.; Wright, M.; Wheller, R.; Nelson, R.; et al. Glu-C, an alternative digestive enzyme for the quantitative LC-MS/MS analysis of an IgG-based antibody biotherapeutic. Bioanalysis 2018, 10, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Kori, Y.; Patel, R.; Neill, A.; Liu, H. A conventional procedure to reduce Asn deamidation artifacts during trypsin peptide mapping. J. Chromatogr. B 2016, 1009–1010, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kolsrud, H.; Malerod, H.; Ray, S.; Reubsaet, L.; Lundanes, E.; Greibrokk, T. A Critical Review of Trypsin Digestion for LC-MS Based Proteomics. In Integrative Proteomics; InTech: Aalesund, Norway, 2012. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, X.; Liu, Y.H.; Richardson, D.; Li, H.; Shameem, M.; Yang, X. Simultaneous monitoring of oxidation, deamidation, isomerization, and glycosylation of monoclonal antibodies by liquid chromatography-mass spectrometry method with ultrafast tryptic digestion. MAbs 2016, 8, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Wang, F.; May, K.; Xu, W.; Liu, H. LC-MS analysis of glycopeptides of recombinant monoclonal antibodies by a rapid digestion procedure. J. Chromatogr. B 2012, 907, 87–93. [Google Scholar] [CrossRef]
- Gaza-Bulseco, G.; Li, B.; Bulseco, A.; Liu, H. Method to Differentiate Asn Deamidation That Occurred Prior to and during Sample Preparation of a Monoclonal Antibody. Anal. Chem. 2008, 80, 9491–9498. [Google Scholar] [CrossRef]
- Li, X.; Cournoyer, J.J.; Lin, C.; O’Connor, P.B. Use of 18O Labels to Monitor Deamidation during Protein and Peptide Sample Processing. J. Am. Soc. Mass Spectrom. 2008, 19, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Xu, W.; Niu, B.; Kabundi, I.; Luo, H.; Prophet, M.; Chen, W.; Liu, D.; Saveliev, S.V.; Urh, M.; et al. An Automated and Qualified Platform Method for Site-Specific Succinimide and Deamidation Quantitation Using Low-pH Peptide Mapping. J. Pharm. Sci. 2019, 108, 3540–3549. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Li, F.; Ding, J. Development of an efficient LC-MS peptide mapping method using accelerated sample preparation for monoclonal antibodies. J. Chromatogr. B 2020, 1137, 121895. [Google Scholar] [CrossRef]
- Cingöz, A.; Hugon-Chapuis, F.; Pichon, V. Evaluation of various immobilized enzymatic microreactors coupled on-line with liquid chromatography and mass spectrometry detection for quantitative analysis of cytochrome c. J. Chromatogr. A 2008, 1209, 95–103. [Google Scholar] [CrossRef]
- Goyon, A.; Dai, L.; Chen, T.; Wei, B.; Yang, F.; Andersen, N.; Kopf, R.; Leiss, M.; Mølhøj, M.; Guillarme, D.; et al. From proof of concept to the routine use of an automated and robust multi-dimensional liquid chromatography mass spectrometry workflow applied for the charge variant characterization of therapeutic antibodies. J. Chromatogr. A 2019, 1615, 460740. [Google Scholar] [CrossRef]
- Yin, Z.; Zhao, W.; Tian, M.; Zhang, Q.; Guo, L.; Yang, L. A capillary electrophoresis-based immobilized enzyme reactor using graphene oxide as a support via layer by layer electrostatic assembly. Analyst 2014, 139, 1973–1979. [Google Scholar] [CrossRef]
- Zeisbergerová, M.; Adámková, A.; Glatz, Z. Integration of on-line protein digestion by trypsin in CZE by means of electrophoretically mediated microanalysis. Electrophoresis 2009, 30, 2378–2384. [Google Scholar] [CrossRef]
- Ladner, Y.; Mas, S.; Coussot, G.; Bartley, K.; Montels, J.; Morel, J.; Perrin, C. Integrated microreactor for enzymatic reaction automation: An easy step toward the quality control of monoclonal antibodies. J. Chromatogr. A 2017, 1528, 83–90. [Google Scholar] [CrossRef]
- Guttman, A. Capillary electrophoresis in the N-glycosylation analysis of biopharmaceuticals. TrAC Trends Anal. Chem. 2013, 48, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, J.; Cosgrave, E.F.J.; Allhorn, M.; Nordgren, M.; Björk, S.; Olsson, F.; Fredriksson, S.; Collin, M. EndoS and EndoS2 hydrolyze Fc-glycans on therapeutic antibodies with different glycoform selectivity and can be used for rapid quantification of high-mannose glycans. Glycobiology 2015, 25, 1053–1063. [Google Scholar] [CrossRef] [Green Version]
- Henninot, A.; Terrier, A.; Charton, J.; Urbain, R.; Fontayne, A.; Deprez, B.; Beghyn, T. Characterization of monoclonal antibodies by a fast and easy liquid chromatography-mass spectrometry time-of-flight analysis on culture supernatant. Anal. Biochem. 2015, 491, 52–54. [Google Scholar] [CrossRef]
- Ruhaak, L.R.; Zauner, G.; Huhn, C.; Bruggink, C.; Deelder, A.M.; Wuhrer, M. Glycan labeling strategies and their use in identification and quantification. Anal. Bioanal. Chem. 2010, 397, 3457–3481. [Google Scholar] [CrossRef] [Green Version]
- Danyluk, H.J.; Shum, L.K.; Zandberg, W.F. A rapid procedure for the purification of 8-aminopyrene trisulfonate (APTS)-labeled glycans for capillary electrophoresis (CE)-based enzyme assays. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1588, pp. 223–236. [Google Scholar]
- Váradi, C.; Lew, C.; Guttman, A. Rapid magnetic bead based sample preparation for automated and high throughput N-glycan analysis of therapeutic antibodies. Anal. Chem. 2014, 86, 5682–5687. [Google Scholar] [CrossRef]
- Szigeti, M.; Lew, C.; Roby, K.; Guttman, A. Fully Automated Sample Preparation for Ultrafast N-Glycosylation Analysis of Antibody Therapeutics. J. Lab. Autom. 2016, 21, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Crihfield, C.L.; Gattu, S.; Veltri, L.M.; Holland, L.A. Capillary Electrophoresis Separations of Glycans. Chem. Rev. 2018, 118, 7867–7885. [Google Scholar] [CrossRef] [Green Version]
- Staub, A.; Guillarme, D.; Schappler, J.; Veuthey, J.-L.; Rudaz, S. Intact protein analysis in the biopharmaceutical field. J. Pharm. Biomed. Anal. 2011, 55, 810–822. [Google Scholar] [CrossRef]
- Shi, Y.; Li, Z.; Qiao, Y.; Lin, J. Development and validation of a rapid capillary zone electrophoresis method for determining charge variants of mAb. J. Chromatogr. B 2012, 906, 63–68. [Google Scholar] [CrossRef]
- Goyon, A.; Francois, Y.N.; Colas, O.; Beck, A.; Veuthey, J.L.; Guillarme, D. High-resolution separation of monoclonal antibodies mixtures and their charge variants by an alternative and generic CZE method. Electrophoresis 2018, 39, 2083–2090. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.N.; Petersen, N.J.; Rand, K.D. A simple sheathless CE-MS interface with a sub-micrometer electrical contact fracture for sensitive analysis of peptide and protein samples. Anal. Chim. Acta 2016, 936, 157–167. [Google Scholar] [CrossRef]
- Haselberg, R.; De Vijlder, T.; Heukers, R.; Smit, M.J.; Romijn, E.P.; Somsen, G.W.; Domínguez-Vega, E. Heterogeneity assessment of antibody-derived therapeutics at the intact and middle-up level by low-flow sheathless capillary electrophoresis-mass spectrometry. Anal. Chim. Acta 2018, 1044, 181–190. [Google Scholar] [CrossRef]
- Belov, A.M.; Zang, L.; Sebastiano, R.; Santos, M.R.; Bush, D.R.; Karger, B.L.; Ivanov, A.R. Complementary middle-down and intact monoclonal antibody proteoform characterization by capillary zone electrophoresis—mass spectrometry. Electrophoresis 2018, 39, 2069–2082. [Google Scholar] [CrossRef]
- Maxwell, E.J.; Chen, D.D.Y. Twenty years of interface development for capillary electrophoresis–electrospray ionization–mass spectrometry. Anal. Chim. Acta 2008, 627, 25–33. [Google Scholar] [CrossRef]
- Ramautar, R.; Heemskerk, A.A.M.; Hensbergen, P.J.; Deelder, A.M.; Busnel, J.-M.; Mayboroda, O.A. CE–MS for proteomics: Advances in interface development and application. J. Proteomics 2012, 75, 3814–3828. [Google Scholar] [CrossRef]
- Heemskerk, A.A.M.; Deelder, A.M.; Mayboroda, O.A. CE-ESI-MS for bottom-up proteomics: Advances in separation, interfacing and applications. Mass Spectrom. Rev. 2016, 35, 259–271. [Google Scholar] [CrossRef]
- Kawai, T. Focusing Review Recent Studies on Online Sample Preconcentration Methods in Capillary Electrophoresis Coupled with Mass Spectrometry. Chromatography 2017, 38, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gahoual, R.; Leize-Wagner, E.; Houzé, P.; François, Y.N. Revealing the potential of capillary electrophoresis/mass spectrometry: The tipping point. Rapid Commun. Mass Spectrom. 2019, 33, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Mokaddem, M.; Gareil, P.; Belgaied, J.-E.; Varenne, A. A new insight into suction and dilution effects in capillary electrophoresis coupled to mass spectrometry via an electrospray ionization interface. Part I-Suction effect. Electrophoresis 2008, 29, 1957–1964. [Google Scholar] [CrossRef]
- Höcker, O.; Montealegre, C.; Neusüß, C. Characterization of a nanoflow sheath liquid interface and comparison to a sheath liquid and a sheathless porous-tip interface for CE-ESI-MS in positive and negative ionization. Anal. Bioanal. Chem. 2018, 410, 5265–5275. [Google Scholar] [CrossRef]
- Gahoual, R.; Busnel, J.-M.; Wolff, P.; François, Y.N.; Leize-Wagner, E. Novel sheathless CE-MS interface as an original and powerful infusion platform for nanoESI study: From intact proteins to high molecular mass noncovalent complexes. Anal. Bioanal. Chem. 2014, 406, 1029–1038. [Google Scholar] [CrossRef]
- Whitmore, C.D.; Gennaro, L.A. Capillary electrophoresis-mass spectrometry methods for tryptic peptide mapping of therapeutic antibodies. Electrophoresis 2012, 33, 1550–1556. [Google Scholar] [CrossRef]
- Metwally, H.; McAllister, R.G.; Konermann, L. Exploring the Mechanism of Salt-Induced Signal Suppression in Protein Electrospray Mass Spectrometry Using Experiments and Molecular Dynamics Simulations. Anal. Chem. 2015, 87, 2434–2442. [Google Scholar] [CrossRef]
- Biacchi, M.; Bhajun, R.; Saïd, N.; Beck, A.; François, Y.N.; Leize-Wagner, E. Analysis of monoclonal antibody by a novel CE-UV/MALDI-MS interface. Electrophoresis 2014, 35, 2986–2995. [Google Scholar] [CrossRef] [Green Version]
- Moritz, B.; Schnaible, V.; Kiessig, S.; Heyne, A.; Wild, M.; Finkler, C.; Christians, S.; Mueller, K.; Zhang, L.; Furuya, K.; et al. Evaluation of capillary zone electrophoresis for charge heterogeneity testing of monoclonal antibodies. J. Chromatogr. B 2015, 983–984, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Espinosa- de la Garza, C.E.; Perdomo-Abúndez, F.C.; Padilla-Calderón, J.; Uribe-Wiechers, J.M.; Pérez, N.O.; Flores-Ortiz, L.F.; Medina-Rivero, E. Analysis of recombinant monoclonal antibodies by capillary zone electrophoresis. Electrophoresis 2013, 34, 1133–1140. [Google Scholar] [CrossRef]
- Kahle, J.; Zagst, H.; Wiesner, R.; Wätzig, H. Comparative charge-based separation study with various capillary electrophoresis (CE) modes and cation exchange chromatography (CEX) for the analysis of monoclonal antibodies. J. Pharm. Biomed. Anal. 2019, 147, 460–470. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Isele, C.; Hou, W.; Ruesch, M. Rapid analysis of charge variants of monoclonal antibodies with capillary zone electrophoresis in dynamically coated fused-silica capillary. J. Sep. Sci. 2011, 34, 548–555. [Google Scholar] [CrossRef]
- Jooß, K.; Hühner, J.; Kiessig, S.; Moritz, B.; Neusüß, C. Two-dimensional capillary zone electrophoresis-mass spectrometry for the characterization of intact monoclonal antibody charge variants, including deamidation products. Anal. Bioanal. Chem. 2017, 409, 6057–6067. [Google Scholar] [CrossRef]
- Redman, E.A.; Batz, N.G.; Mellors, J.S.; Ramsey, J.M. Integrated Microfluidic Capillary Electrophoresis-Electrospray Ionization Devices with Online MS Detection for the Separation and Characterization of Intact Monoclonal Antibody Variants. Anal. Chem. 2015, 87, 2264–2272. [Google Scholar] [CrossRef]
- Dadouch, M.; Ladner, Y.; Bich, C.; Larroque, M.; Larroque, C.; Morel, J.; Bonnet, P.A.; Perrin, C. An in-line enzymatic microreactor for the middle-up analysis of monoclonal antibodies by capillary electrophoresis. Analyst 2020, 145, 1759–1767. [Google Scholar] [CrossRef]
- Henley, W.H.; He, Y.; Mellors, J.S.; Batz, N.G.; Ramsey, J.M.; Jorgenson, J.W. High resolution separations of charge variants and disulfide isomers of monoclonal antibodies and antibody drug conjugates using ultra-high voltage capillary electrophoresis with high electric field strength. J. Chromatogr. A 2017, 1523, 72–79. [Google Scholar] [CrossRef]
- Suba, D.; Urbányi, Z.; Salgó, A. Method development and qualification of capillary zone electrophoresis for investigation of therapeutic monoclonal antibody quality. J. Chromatogr. B 2016, 1032, 224–229. [Google Scholar] [CrossRef]
- Moritz, B.; Locatelli, V.; Niess, M.; Bathke, A.; Kiessig, S.; Entler, B.; Finkler, C.; Wegele, H.; Stracke, J. Optimization of capillary zone electrophoresis for charge heterogeneity testing of biopharmaceuticals using enhanced method development principles. Electrophoresis 2017, 38, 3136–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biacchi, M.; Gahoual, R.; Said, N.; Beck, A.; Leize-Wagner, E.; François, Y.-N. Glycoform Separation and Characterization of Cetuximab Variants by Middle-up Off-Line Capillary Zone Electrophoresis-UV/Electrospray Ionization-MS. Anal. Chem. 2015, 87, 6240–6250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biacchi, M.; Said, N.; Beck, A.; Leize-Wagner, E.; François, Y.-N. Top-down and middle-down approach by fraction collection enrichment using off-line capillary electrophoresis—mass spectrometry coupling: Application to monoclonal antibody Fc/2 charge variants. J. Chromatogr. A 2017, 1498, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Sun, L.; Knierman, M.D.; Dovichi, N.J. Fast separation and analysis of reduced monoclonal antibodies with capillary zone electrophoresis coupled to mass spectrometry. Talanta 2016, 148, 529–533. [Google Scholar] [CrossRef]
- Redman, E.A.; Mellors, J.S.; Starkey, J.A.; Ramsey, J.M. Characterization of Intact Antibody Drug Conjugate Variants Using Microfluidic Capillary Electrophoresis-Mass Spectrometry. Anal. Chem. 2016, 88, 2220–2226. [Google Scholar] [CrossRef]
- Han, M.; Rock, B.M.; Pearson, J.T.; Rock, D.A. Intact mass analysis of monoclonal antibodies by capillary electrophoresis—Mass spectrometry. J. Chromatogr. B 2016, 1011, 24–32. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, L.; Champion, M.M.; Knierman, M.D.; Dovichi, N.J. Capillary Zone Electrophoresis–Electrospray Ionization-Tandem Mass Spectrometry for Top-Down Characterization of the Mycobacterium marinum Secretome. Anal. Chem. 2014, 86, 4873–4878. [Google Scholar] [CrossRef]
- Giorgetti, J.; Lechner, A.; Del Nero, E.; Beck, A.; François, Y.N.; Leize-Wagner, E. Intact monoclonal antibodies separation and analysis by sheathless capillary electrophoresis-mass spectrometry. Eur. J. Mass Spectrom. 2019, 25, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, J.; Beck, A.; Leize-Wagner, E.; François, Y.N. Combination of intact, middle-up and bottom-up levels to characterize 7 therapeutic monoclonal antibodies by capillary electrophoresis—Mass spectrometry. J. Pharm. Biomed. Anal. 2020, 182, 113107. [Google Scholar] [CrossRef] [Green Version]
- Gennaro, L.A.; Salas-Solano, O. On-Line CE−LIF−MS Technology for the Direct Characterization of N-Linked Glycans from Therapeutic Antibodies. Anal. Chem. 2008, 80, 3838–3845. [Google Scholar] [CrossRef]
- Ambrogelly, A.; Gozo, S.; Katiyar, A.; Dellatore, S.; Kune, Y.; Bhat, R.; Sun, J.; Li, N.; Wang, D.; Nowak, C.; et al. Analytical comparability study of recombinant monoclonal antibody therapeutics. MAbs 2018, 10, 513–538. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Gahoual, R.; Kuhn, L.; Beck, A.; François, Y.-N.; Leize-Wagner, E. Structural characterization of antibody drug conjugate by a combination of intact, middle-up and bottom-up techniques using sheathless capillary electrophoresis—Tandem mass spectrometry as nanoESI infusion platform and separation method. Anal. Chim. Acta 2016, 918, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carillo, S.; Jakes, C.; Bones, J. In-depth analysis of monoclonal antibodies using microfluidic capillary electrophoresis and native mass spectrometry. J. Pharm. Biomed. Anal. 2020, 185, 113218. [Google Scholar] [CrossRef] [PubMed]
- Belov, A.M.; Viner, R.; Santos, M.R.; Horn, D.M.; Bern, M.; Karger, B.L.; Ivanov, A.R. Analysis of Proteins, Protein Complexes, and Organellar Proteomes Using Sheathless Capillary Zone Electrophoresis—Native Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 2614–2634. [Google Scholar] [CrossRef] [PubMed]
- Le-Minh, V.; Tran, N.T.; Makky, A.; Rosilio, V.; Taverna, M.; Smadja, C. Capillary zone electrophoresis-native mass spectrometry for the quality control of intact therapeutic monoclonal antibodies. J. Chromatogr. A 2019, 1601, 375–384. [Google Scholar] [CrossRef]
- Zhang, H.; Ge, Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ. Cardiovasc. Genet. 2011, 4, 711. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, L.A.; Salas-Solano, O.; Ma, S. Capillary electrophoresis–mass spectrometry as a characterization tool for therapeutic proteins. Anal. Biochem. 2006, 355, 249–258. [Google Scholar] [CrossRef]
- Gahoual, R.; Busnel, J.-M.; Beck, A.; François, Y.-N.; Leize-Wagner, E. Full Antibody Primary Structure and Microvariant Characterization in a Single Injection Using Transient Isotachophoresis and Sheathless Capillary Electrophoresis–Tandem Mass Spectrometry. Anal. Chem. 2014, 86, 9074–9081. [Google Scholar] [CrossRef] [Green Version]
- Gahoual, R.; Biacchi, M.; Chicher, J.; Kuhn, L.; Hammann, P.; Beck, A.; Leize-Wagner, E.; François, Y.N. Monoclonal antibodies biosimilarity assessment using transient isotachophoresis capillary zone electrophoresis-tandem mass spectrometry. MAbs 2014, 6, 1464–1473. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, J.; D’Atri, V.; Canonge, J.; Lechner, A.; Guillarme, D.; Colas, O.; Wagner-Rousset, E.; Beck, A.; Leize-Wagner, E.; François, Y.-N. Monoclonal antibody N-glycosylation profiling using capillary electrophoresis—Mass spectrometry: Assessment and method validation. Talanta 2018, 178, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Janin-Bussat, M.-C.; Dillenbourg, M.; Corvaia, N.; Beck, A.; Klinguer-Hamour, C. Characterization of antibody drug conjugate positional isomers at cysteine residues by peptide mapping LC-MS analysis. J. Chromatogr. B 2015, 981–982, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Štěpánová, S.; Kašička, V. Recent applications of capillary electromigration methods to separation and analysis of proteins. Anal. Chim. Acta 2016, 933, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, A.A.M.; Wuhrer, M.; Busnel, J.-M.; Koeleman, C.A.M.; Selman, M.H.J.; Vidarsson, G.; Kapur, R.; Schoenmaker, B.; Derks, R.J.E.; Deelder, A.M.; et al. Coupling porous sheathless interface MS with transient-ITP in neutral capillaries for improved sensitivity in glycopeptide analysis. Electrophoresis 2013, 34, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Gahoual, R.; Beck, A.; François, Y.-N.; Leize-Wagner, E. Independent highly sensitive characterization of asparagine deamidation and aspartic acid isomerization by sheathless CZE-ESI-MS/MS. J. Mass Spectrom. 2016, 51, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Kammeijer, G.S.M.; Jansen, B.C.; Kohler, I.; Heemskerk, A.A.M.; Mayboroda, O.A.; Hensbergen, P.J.; Schappler, J.; Wuhrer, M. Sialic acid linkage differentiation of glycopeptides using capillary electrophoresis—Electrospray ionization—Mass spectrometry. Sci. Rep. 2017, 7, 3733. [Google Scholar] [CrossRef]
- Dada, O.O.; Zhao, Y.; Jaya, N.; Salas-Solano, O. High-Resolution Capillary Zone Electrophoresis with Mass Spectrometry Peptide Mapping of Therapeutic Proteins: Improved Separation with Mixed Aqueous–Aprotic Dipolar Solvents (N, N-Dimethylacetamide and N, N-Dimethylformamide) as the Background Electrolyte. Anal. Chem. 2017, 89, 11227–11235. [Google Scholar] [CrossRef]
- Dada, O.O.; Zhao, Y.; Jaya, N.; Salas-Solano, O. High-Resolution Capillary Zone Electrophoresis with Mass Spectrometry Peptide Mapping of Therapeutic Proteins: Peptide Recovery and Post-translational Modification Analysis in Monoclonal Antibodies and Antibody-Drug Conjugates. Anal. Chem. 2017, 89, 11236–11242. [Google Scholar] [CrossRef]
- Bongers, J.; Cummings, J.J.; Ebert, M.B.; Federici, M.M.; Gledhill, L.; Gulati, D.; Hilliard, G.M.; Jones, B.H.; Lee, K.R.; Mozdzanowski, J.; et al. Validation of a peptide mapping method for a therapeutic monoclonal antibody: What could we possibly learn about a method we have run 100 times? J. Pharm. Biomed. Anal. 2000, 21, 1099–1128. [Google Scholar] [CrossRef]
- Krylova, S.M.; Okhonin, V.; Krylov, S.N. Transverse diffusion of laminar flow profiles—A generic method for mixing reactants in capillary microreactor. J. Sep. Sci. 2009, 32, 742–756. [Google Scholar] [CrossRef]
- Ladner, Y.; Mas, S.; Coussot, G.; Montels, J.; Perrin, C. In-line tryptic digestion of therapeutic molecules by capillary electrophoresis with temperature control. Talanta 2019, 193, 146–151. [Google Scholar] [CrossRef]
- Zhang, L.; Luo, S.; Zhang, B. Glycan analysis of therapeutic glycoproteins. MAbs 2016, 8, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittermayr, S.; Bones, J.; Guttman, A. Unraveling the glyco-puzzle: Glycan structure identification by capillary electrophoresis. Anal. Chem. 2013, 85, 4228–4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Santos, M.; Guttman, A. Comparative core fucosylation analysis of some major therapeutic antibody N-glycans by direct infusion ESI-MS and CE-LIF detection. J. Sep. Sci. 2013, 36, 2862–2867. [Google Scholar] [CrossRef] [PubMed]
- Maeda, E.; Kita, S.; Kinoshita, M.; Urakami, K.; Hayakawa, T.; Kakehi, K. Analysis of nonhuman N-glycans as the minor constituents in recombinant monoclonal antibody pharmaceuticals. Anal. Chem. 2012, 84, 2373–2379. [Google Scholar] [CrossRef]
- Bunz, S.C.; Cutillo, F.; Neusüß, C. Analysis of native and APTS-labeled N-glycans by capillary electrophoresis/time-of-flight mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 8277–8284. [Google Scholar] [CrossRef] [PubMed]
- Bunz, S.-C.; Rapp, E.; Neusüss, C. Capillary Electrophoresis/Mass Spectrometry of APTS-Labeled Glycans for the Identification of Unknown Glycan Species in Capillary Electrophoresis/Laser-Induced Fluorescence Systems. Anal. Chem. 2013, 85, 10218–10224. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Su, M.; Rifai, F.N.; Li, P.; Li, S.F.Y. Parallel analysis and orthogonal identification of N-glycans with different capillary electrophoresis mechanisms. Anal. Chim. Acta 2017, 953, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Guttman, A.; Bones, J.; Karger, B.L. Rapid high-resolution characterization of functionally important monoclonal antibody N -glycans by capillary electrophoresis. Anal. Chem. 2011, 83, 5329–5336. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Liu, J.; Szabo, Z.; Kunnummal, B.; Han, X.; Ouyang, Y.; Linhardt, R.J.; Xia, Q. On-line capillary electrophoresis/laser-induced fluorescence/mass spectrometry analysis of glycans labeled with TealTM fluorescent dye using an electrokinetic sheath liquid pump-based nanospray ion source. Rapid Commun. Mass Spectrom. 2018, 32, 882–888. [Google Scholar] [CrossRef]
- Váradi, C.; Mittermayr, S.; Millán-Martín, S.; Bones, J. Quantitative twoplex glycan analysis using 12C6 and 13C6 stable isotope 2-aminobenzoic acid labelling and capillary electrophoresis mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 8691–8700. [Google Scholar] [CrossRef]
- Váradi, C.; Jakes, C.; Bones, J. Analysis of Cetuximab N-Glycosylation using Multiple Fractionation Methods and Capillary Electrophoresis Mass Spectrometry. J. Pharm. Biomed. Anal. 2019, 182, 113035. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Archer-Hartmann, S.A.; Holland, L.A. Transformable capillary electrophoresis for oligosaccharide separations using phospholipid additives. Anal. Chem. 2010, 82, 1228–1233. [Google Scholar] [CrossRef]
- Archer-Hartmann, S.A.; Crihfield, C.L.; Holland, L.A. Online enzymatic sequencing of glycans from Trastuzumab by phospholipid-assisted capillary electrophoresis. Electrophoresis 2011, 32, 3491–3498. [Google Scholar] [CrossRef] [PubMed]
- Jooß, K.; Meckelmann, S.W.; Klein, J.; Schmitz, O.J.; Neusüß, C. Capillary zone electrophoresis coupled to drift tube ion mobility-mass spectrometry for the analysis of native and APTS-labeled N-glycans. Anal. Bioanal. Chem. 2019, 411, 6255–6264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Albanetti, T.; Linkous, T.; Larkin, C.J.; Schoner, R.; McGivney, J.B.; Dovichi, N.J. Comprehensive analysis of host cell impurities in monoclonal antibodies with improved sensitivity by capillary zone electrophoresis mass spectrometry. Electrophoresis 2017, 38, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Sun, L.; Linkous, T.; Kernaghan, D.; Mcgivney, J.B.; Dovichi, N.J. Absolute quantitation of host cell proteins in recombinant human monoclonal antibodies with an automated CZE-ESI-MS/MS system. Electrophoresis 2014, 35, 1448–1452. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Sun, L.; Heidbrink-Thompson, J.; Kuntumalla, S.; Lin, H.Y.; Larkin, C.J.; Mcgivney, J.B.; Dovichi, N.J. Capillary zone electrophoresis tandem mass spectrometry detects low concentration host cell impurities in monoclonal antibodies. Electrophoresis 2016, 37, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Chen, Z.; Snovida, S.; Liu, Y.; Rogers, J.C.; Li, L. Capillary Electrophoresis-Electrospray Ionization-Mass Spectrometry for Quantitative Analysis of Glycans Labeled with Multiplex Carbonyl-Reactive Tandem Mass Tags. Anal. Chem. 2015, 87, 6527–6534. [Google Scholar] [CrossRef]
- Huhn, C.; Ruhaak, L.R.; Mannhardt, J.; Wuhrer, M.; Neusüß, C.; Deelder, A.M.; Meyer, H. Alignment of laser-induced fluorescence and mass spectrometric detection traces using electrophoretic mobility scaling in CE-LIF-MS of labeled N-glycans. Electrophoresis 2012, 33, 563–566. [Google Scholar] [CrossRef]
- Szekrényes, Á.; Park, S.A.S.; Santos, M.; Lew, C.; Jones, A.; Haxo, T.; Kimzey, M.; Pourkaveh, S.; Szabó, Z.; Sosic, Z.; et al. Multi-site N-glycan mapping study 1: Capillary electrophoresis—Laser induced fluorescence. MAbs 2016, 8, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Rustandi, R.R.; Washabaugh, M.W.; Wang, Y. Applications of CE SDS gel in development of biopharmaceutical antibody-based products. Electrophoresis 2008, 29, 3612–3620. [Google Scholar] [CrossRef] [PubMed]
- Kotia, R.B.; Raghani, A.R. Analysis of monoclonal antibody product heterogeneity resulting from alternate cleavage sites of signal peptide. Anal. Biochem. 2010, 399, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Lacher, N.A.; Roberts, R.K.; He, Y.; Cargill, H.; Kearns, K.M.; Holovics, H.; Ruesch, M.N. Development, validation, and implementation of capillary gel electrophoresis as a replacement for SDS-PAGE for purity analysis of IgG2 mAbs. J. Sep. Sci. 2010, 33, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Burman, S.; Gunturi, S.; Foley, J.P. Method development and validation of capillary sodium dodecyl sulfate gel electrophoresis for the characterization of a monoclonal antibody. J. Pharm. Biomed. Anal. 2010, 53, 1236–1243. [Google Scholar] [CrossRef]
- Sänger van de Griend, C.E. CE-SDS method development, validation, and best practice—An overview. Electrophoresis 2019, 40, 2361–2374. [Google Scholar] [CrossRef]
- Beckman, J.; Song, Y.; Gu, Y.; Voronov, S.; Chennamsetty, N.; Krystek, S.; Mussa, N.; Li, Z.J. Purity Determination by Capillary Electrophoresis Sodium Hexadecyl Sulfate (CE-SHS): A Novel Application for Therapeutic Protein Characterization. Anal. Chem. 2018, 90, 2542–2547. [Google Scholar] [CrossRef]
- Turner, A.; Yandrofski, K.; Telikepalli, S.; King, J.; Heckert, A.; Filliben, J.; Ripple, D.; Schiel, J.E. Development of orthogonal NISTmAb size heterogeneity control methods. Anal. Bioanal. Chem. 2018, 410, 2095–2110. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Li, W.; Zhao, H.; Wang, A.; Lei, Y.; Xie, Q.; Xiong, S. Discovery and characterization of CHO host cell protease-induced fragmentation of a recombinant monoclonal antibody during production process development. J. Chromatogr. B 2019, 1112, 1–10. [Google Scholar] [CrossRef]
- Hutterer, K.M.; Hong, R.W.; Lull, J.; Zhao, X.; Wang, T.; Pei, R.; Le, M.E.; Borisov, O.; Piper, R.; Liu, Y.D.; et al. Monoclonal antibody disulfide reduction during manufacturing. MAbs 2013, 5, 608–613. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Liu, Y.D.; Cai, B.; Huang, G.; Flynn, G.C. Investigation of antibody disulfide reduction and re-oxidation and impact to biological activities. J. Pharm. Biomed. Anal. 2015, 102, 519–528. [Google Scholar] [CrossRef]
- Guo, A.; Han, M.; Martinez, T.; Ketchem, R.R.; Novick, S.; Jochheim, C.; Balland, A. Electrophoretic evidence for the presence of structural isoforms specific for the IgG2 isotype. Electrophoresis 2008, 29, 2550–2556. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, Z.; Lin, J. Advantages of CE-SDS over SDS-PAGE in mAb purity analysis. Anal. Methods 2012, 4, 1637–1642. [Google Scholar] [CrossRef]
- Szekrényes, Á.; Roth, U.; Kerékgyártó, M.; Székely, A.; Kurucz, I.; Kowalewski, K.; Guttman, A. High-throughput analysis of therapeutic and diagnostic monoclonal antibodies by multicapillary SDS gel electrophoresis in conjunction with covalent fluorescent labeling. Anal. Bioanal. Chem. 2012, 404, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.T.; Zhang, S.; Adams, T.; DiPaolo, B.; Dally, J. Establishment and validation of a microfluidic capillary gel electrophoresis platform method for purity analysis of therapeutic monoclonal antibodies. Electrophoresis 2017, 38, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lu, J.J.; Liu, S. Protein separation by capillary gel electrophoresis: A review. Anal. Chim. Acta 2012, 709, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Römer, J.; Montealegre, C.; Schlecht, J.; Kiessig, S.; Moritz, B.; Neusüß, C. Online mass spectrometry of CE (SDS)-separated proteins by two-dimensional capillary electrophoresis. Anal. Bioanal. Chem. 2019, 411, 7197–7206. [Google Scholar] [CrossRef]
- Wang, W.H.; Cheung-Lau, J.; Chen, Y.; Lewis, M.; Tang, Q.M. Specific and high-resolution identification of monoclonal antibody fragments detected by capillary electrophoresis–sodium dodecyl sulfate using reversed-phase HPLC with top-down mass spectrometry analysis. MAbs 2019, 11, 1233–1244. [Google Scholar] [CrossRef]
- Sánchez-Hernández, L.; Montealegre, C.; Kiessig, S.; Moritz, B.; Neusüß, C. In-capillary approach to eliminate SDS interferences in antibody analysis by capillary electrophoresis coupled to mass spectrometry. Electrophoresis 2017, 38, 1044–1052. [Google Scholar] [CrossRef]
- Borza, B.; Szigeti, M.; Szekrenyes, A.; Hajba, L.; Guttman, A. Glycosimilarity assessment of biotherapeutics 1: Quantitative comparison of the N-glycosylation of the innovator and a biosimilar version of etanercept. J. Pharm. Biomed. Anal. 2018, 153, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Reusch, D.; Haberger, M.; Maier, B.; Maier, M.; Kloseck, R.; Zimmermann, B.; Hook, M.; Szabo, Z.; Tep, S.; Wegstein, J.; et al. Comparison of methods for the analysis of therapeutic immunoglobulin G Fc-glycosylation profiles—Part 1: Separation-based methods. MAbs 2015, 7, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Jarvas, G.; Szigeti, M.; Guttman, A. Structural identification of N-linked carbohydrates using the GUcal application: A tutorial. J. Proteomics 2018, 171, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Filep, C.; Borza, B.; Jarvas, G.; Guttman, A. N-glycosylation analysis of biopharmaceuticals by multicapillary gel electrophoresis: Generation and application of a new glucose unit database. J. Pharm. Biomed. Anal. 2020, 178, 112892. [Google Scholar] [CrossRef] [PubMed]
- Behne, A.; Muth, T.; Borowiak, M.; Reichl, U.; Rapp, E. glyXalign: High-throughput migration time alignment preprocessing of electrophoretic data retrieved via multiplexed capillary gel electrophoresis with laser-induced fluorescence detection-based glycoprofiling. Electrophoresis 2013, 34, 2311–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.; Colas, O.; Chenu, S.; Goyon, A.; Murisier, A.; Cianferani, S.; François, Y.; Fekete, S.; Guillarme, D.; D’Atri, V.; et al. Determination of size variants by CE-SDS for approved therapeutic antibodies: Key implications of subclasses and light chain specificities. J. Pharm. Biomed. Anal. 2020, 184, 113166. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yu, C.; Wang, W.; Wang, L. Interlaboratory method validation of icIEF methodology for analysis of monoclonal antibodies. Electrophoresis 2018, 39, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Duhamel, L.; Gu, Y.; Barnett, G.; Tao, Y.; Voronov, S.; Ding, J.; Mussa, N.; Li, Z.J. Therapeutic protein purity and fragmented species characterization by capillary electrophoresis sodium dodecyl sulfate using systematic hybrid cleavage and forced degradation. Anal. Bioanal. Chem. 2019, 411, 5617–5629. [Google Scholar] [CrossRef]
- Kerékgyártó, M.; Guttman, A. Toward the generation of an aminonaphthalene trisulfonate labeled N -glycan database for capillary gel electrophoresis analysis of carbohydrates. Electrophoresis 2014, 35, 2222–2228. [Google Scholar] [CrossRef]
- Goyon, A.; Excoffier, M.; Janin-Bussat, M.-C.; Bobaly, B.; Fekete, S.; Guillarme, D.; Beck, A. Determination of isoelectric points and relative charge variants of 23 therapeutic monoclonal antibodies. J. Chromatogr. B. 2017, 1065–1066, 119–128. [Google Scholar] [CrossRef]
- Michels, D.A.; Salas-Solano, O.; Felten, C. Imaged Capillary Isoelectric Focusing for Charge-Variant Analysis of Biopharmaceuticals. BioProcess Int. 2011, 9, 48–54. [Google Scholar]
- Zhu, G.; Sun, L.; Wojcik, R.; Kernaghan, D.; McGivney, J.B.; Dovichi, N.J. A rapid cIEF–ESI–MS/MS method for host cell protein analysis of a recombinant human monoclonal antibody. Talanta 2012, 98, 253–256. [Google Scholar] [CrossRef]
- Shimura, K. Capillary Isoelectric Focusing. In Capillary Electromigration Separation Methods; Elsevier: Amsterdam, The Netherlands, 2018; pp. 167–187. [Google Scholar] [CrossRef]
- Hühner, J.; Neusüß, C. CIEF-CZE-MS applying a mechanical valve. Anal. Bioanal. Chem. 2016, 408, 4055–4061. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, T. Peak identification in capillary isoelectric focusing using the concept of relative peak position as determined by two isoelectric point markers. Electrophoresis 2006, 27, 3584–3590. [Google Scholar] [CrossRef] [PubMed]
- Suba, D.; Urbányi, Z.; Salgó, A. Capillary isoelectric focusing method development and validation for investigation of recombinant therapeutic monoclonal antibody. J. Pharm. Biomed. Anal. 2015, 114, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Tan, Q.; Wang, S. A high-resolution capillary isoelectric focusing method for the determination of therapeutic recombinant monoclonal antibody. J. Sep. Sci. 2011, 34, 1696–1702. [Google Scholar] [CrossRef]
- He, X.Z.; Que, A.H.; Mo, J.J. Analysis of charge heterogeneities in mAbs using imaged CE. Electrophoresis 2009, 30, 714–722. [Google Scholar] [CrossRef]
- Mokaddem, M.; d’Orlyé, F.; Varenne, A. Online Capillary IsoElectric Focusing-ElectroSpray Ionization Mass Spectrometry (CIEF-ESI MS) in Glycerol—Water Media for the Separation and Characterization of Hydrophilic and Hydrophobic Proteins; Humana Press: New York, NY, USA, 2016; pp. 57–66. [Google Scholar]
- Dai, J.; Lamp, J.; Xia, Q.; Zhang, Y. Capillary Isoelectric Focusing-Mass Spectrometry Method for the Separation and Online Characterization of Intact Monoclonal Antibody Charge Variants. Anal. Chem. 2018, 90, 2246–2254. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Zhang, Y. A Middle-Up Approach with Online Capillary Isoelectric Focusing/Mass Spectrometry for In-Depth Characterization of Cetuximab Charge Heterogeneity. Anal. Chem. 2018, 90, 14527–14534. [Google Scholar] [CrossRef]
- Salas-Solano, O.; Babu, K.; Park, S.S.; Zhang, X.; Zhang, L.; Sosic, Z.; Boumajny, B.; Zeng, M.; Cheng, K.C.; Reed-Bogan, A.; et al. Intercompany study to evaluate the robustness of capillary isoelectric focusing technology for the analysis of monoclonal antibodies. Chromatographia 2011, 73, 1137–1144. [Google Scholar] [CrossRef]
- Vanam, R.P.; Schneider, M.A.; Marlow, M.S. Rapid quantitative analysis of monoclonal antibody heavy and light chain charge heterogeneity. MAbs 2015, 7, 1118–1127. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Zhang, Y.; Mueller, H.-M.; Shameem, M.; Chen, X. A new tool for monoclonal antibody analysis. MAbs 2014, 6, 879–893. [Google Scholar] [CrossRef]
- Zhang, Z.; Perrault, R.; Zhao, Y.; Ding, J. SpeB proteolysis with imaged capillary isoelectric focusing for the characterization of domain-specific charge heterogeneities of reference and biosimilar Rituximab. J. Chromatogr. B 2016, 1020, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Štěpánová, S.; Kašička, V. Application of Capillary Electromigration Methods for Physicochemical Measurements. In Capillary Electromigration Separation Methods; Elsevier: Amsterdam, The Netherlands, 2018; pp. 547–591. [Google Scholar] [CrossRef]
- Turner, A.; Schiel, J.E. Qualification of NISTmAb charge heterogeneity control assays. Anal. Bioanal. Chem. 2018, 410, 2079–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chemmalil, L.; Ding, J.; Mussa, N.; Li, Z. Imaged capillary isoelectric focusing in native condition: A novel and successful example. Anal. Biochem. 2017, 537, 13–19. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Patel, R.; Ponniah, G.; Nowak, C.; Neill, A.; Gu, Z.; Liu, H. Characterization of recombinant monoclonal antibody variants detected by hydrophobic interaction chromatography and imaged capillary isoelectric focusing electrophoresis. J. Chromatogr. B 2018, 1085, 96–103. [Google Scholar] [CrossRef]
- Zhang, X.; Voronov, S.; Mussa, N.; Li, Z. A novel reagent significantly improved assay robustness in imaged capillary isoelectric focusing. Anal. Biochem. 2017, 521, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Sun, W.; Gong, F.; Liu, W. Charge profiling and stability testing of biosimilar by capillary isoelectric focusing. Electrophoresis 2014, 35, 1461–1468. [Google Scholar] [CrossRef]
- Demirdirek, B.; Lan, W.; Qiu, D.; Ding, W.; Iyer, L.K.; Bolgar, M.S.; Valente, J.J. Comparison of imaged capillary isoelectric focusing and cation exchange chromatography for monitoring dextrose-mediated glycation of monoclonal antibodies in infusion solutions. J. Chromatogr. B 2019, 1105, 156–163. [Google Scholar] [CrossRef]
- Maeda, E.; Urakami, K.; Shimura, K.; Kinoshita, M.; Kakehi, K. Charge heterogeneity of a therapeutic monoclonal antibody conjugated with a cytotoxic antitumor antibiotic, calicheamicin. J. Chromatogr. A 2010, 1217, 7164–7171. [Google Scholar] [CrossRef]
- Kahle, J.; Stein, M.; Wätzig, H. Design of experiments as a valuable tool for biopharmaceutical analysis with (imaged) capillary isoelectric focusing. Electrophoresis 2019, 40, 2382–2389. [Google Scholar] [CrossRef]
- Kinoshita, M.; Nakatsuji, Y.; Suzuki, S.; Hayakawa, T.; Kakehi, K. Quality assurance of monoclonal antibody pharmaceuticals based on their charge variants using microchip isoelectric focusing method. J. Chromatogr. A 2013, 1309, 76–83. [Google Scholar] [CrossRef]
- Mack, S.; Arnold, D.; Bogdan, G.; Bousse, L.; Danan, L.; Dolnik, V.; Ducusin, M.A.; Gwerder, E.; Herring, C.; Jensen, M.; et al. A novel microchip-based imaged CIEF-MS system for comprehensive characterization and identification of biopharmaceutical charge variants. Electrophoresis 2019, 40, 3084–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chen, D.D.Y. Analysis of four therapeutic monoclonal antibodies by online capillary isoelectric focusing directly coupled to quadrupole time-of-flight mass spectrometry. Electrophoresis 2019, 40, 2899–2907. [Google Scholar] [CrossRef] [PubMed]
- Kats, M.; Richberg, P.C.; Hughes, D.E. pH-dependent isoform transitions of a monoclonal antibody monitored by micellar electrokinetic capillary chromatography. Anal. Chem. 1997, 69, 338–343. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CZE for mAbs Analyses | References | |
| Sample preparation | Monoclonal antibodies (mAb) prepared following the approaches (intact, middle-up, or bottom-up) as detailed in Section 2.2, desalted or non-desalted | |
| Sample buffer | Water, mixture acid-organic solvent, ammonium acetate, tris | [34,73,75,89,104] |
| Capillary coating | Uncoated (bottom-up analysis) | [34,114,116,122,123] |
| Neutral coated (e.g., HPMC, PEO, LPA) | [24,62,75,91,97,99] | |
| Positively charged (e.g., c-PEI, M7C4I) | [76,104,105] | |
| BGE |
| [89,91,96,97,98] |
| [34,75,93,94,100,104,105,109,111,123] | |
| Injection | Hydrodynamic | |
| UV detection | 214 nm | [73,89,91,94] |
| MS detection | Online ESI-MS (Sheath liquid or sheathless interface) | [34,102,104,108] |
| Off-line MALDI-MS | [24,98,99] | |
| CZE for Released Glycans Analyses | References | |
| Sample preparation | Release and labeling N-glycans (APTS or 2-AA) (Section 2.2.3.) | |
| Internal standard: glycan ladder (dextran), oligomers of glycans | ||
| Capillary | Uncoated capillary | [131,144] |
| Neutral coated capillary (polyvinyl alcohol (PVA)) | [132,133] | |
| BGE | 6-aminocaproic acid/ammonia (+additives) | [131] |
| Ammonium acetate, ammonium, acetic acid, formic acid | [121,133,136,144,145] | |
| Injection | Hydrodynamic | |
| LIF detection | APTS labeled N-glycans (excitation 488 nm, emission 520 nm) | [131,132,133,139] |
| 2-AA labeled N-glycans (excitation 325 nm, emission 405 nm) | [130,136,145] | |
| MS detection | Online ESI-MS (Sheath liquid or sheathless interface) | |
| CGE–SDS Analysis of mAbs | References | |
| Sample preparation | Intact mAb, deglycosylated mAb, reduced mAb, digested mAb (e.g., IdeS, IgGdE), desalted or non-desalted | |
| ± Fluorescent dye | [64] | |
| ± Internal standard | [164,171,172] | |
| Sample buffer |
| [150,151,158,164] |
| Capillary | Neutral coating (static or dynamic) | |
| Buffer containing a certain amount of SDS as a tris-borate buffer with 0.1% SDS pH 8.45 | [160] | |
| Sieving matrix | Buffer containing 100 mM Tris-HCl, pH 9.0, 1% SDS | [164] |
| Sieving matrix SDS–MW gel buffer (Beckman Coulter), BioRad buffer (BioRad Laboratories), SDS–MW gel buffer (Sciex) | [149] | |
| Injection | Electrokinetic | |
| UV detection | 220 or 280 nm | |
| LIF detection | 3-(2-furoyl)quinoline-2-carboxaldehyde (FQ) label (excitation 488 nm, emission 600 nm) | [118] |
| CGE Analysis of Released Glycans Analyses | References | |
| Sample preparation | Release and labeling N-glycans (with APTS or ANTS) (Section 2.2.4) | |
| Internal standard: glycan ladder (dextran), oligomers of glycans | [146] | |
| Capillary | Silica capillary or neutral coated capillary | [147] |
| Filled with gel, e.g., | ||
| [146] [166] [133] | |
| Prefilled devices, e.g., | ||
| [173] [169] | |
| Injection | Hydrodynamic or electrokinetic | |
| LIF detection | APTS labeled N-glycans (excitation 488 nm, emission 520 nm) | [160,166] |
| ANTS labeled N-glycans (excitation 420 nm, emission 530 nm) | [167] | |
| cIEF-UV and icIEF-UV Analysis of mAbs | References | |
| Sample preparation | Intact mAb, deglycosylated mAb, reduced mAb, digested mAb (e.g., IdeS, SpeB), desalted or non-desalted. | With stabilizer [181,182,184,188,195,199] Without stabilizer [171,182,184,189] |
| Sample buffer | contains the following components: cIEF gel based on cellulose derivative (0.1–0.4% m/v) ± urea (2–7 M) or formamide; Carrier ampholyte or pharmalytes at one range or different ranges of pI; Anolyte stabilizer (L-argininie) and catholyte stabilizer (iminodiacetic acid); pI markers mixture based on peptides or synthetic molecules; | |
| Capillary | Neutral coated capillaries filled with the sample matrix | |
| BGE | Anolyte: sodium hydroxide ± cellulose derivative Catholyte: phosphoric acid ± cellulose derivative | |
| Injection | Hydrodynamic | |
| UV detection | 280 nm | |
| cIEF-MS Analysis of mAbs | References | |
| Sample preparation | Intact mAb, deglycosylated mAb, reduced mAb, digested mAb (e.g., IdeS), desalted. | [184,185,201] |
| Sample buffer | contains the following components: Glycerol (5–20%); Carrier ampholyte or pharmalyte at one range or different ranges of pI; ± Anolyte stabilizer (L-arginine) and catholyte stabilizer (iminodiacetic acid); pI markers mixture based on peptides or synthetic molecules | |
| Capillary | Neutral coated capillaries filled with the sample matrix | |
| BGE | Anolyte: ammonium hydroxide + glycerol Catholyte: formic acid or acetic acid + glycerol | |
| Injection | Hydrodynamic | |
| MS detection | Online ESI-MS (Sheath liquid, flow-through microvial interface) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dadouch, M.; Ladner, Y.; Perrin, C. Analysis of Monoclonal Antibodies by Capillary Electrophoresis: Sample Preparation, Separation, and Detection. Separations 2021, 8, 4. https://doi.org/10.3390/separations8010004
Dadouch M, Ladner Y, Perrin C. Analysis of Monoclonal Antibodies by Capillary Electrophoresis: Sample Preparation, Separation, and Detection. Separations. 2021; 8(1):4. https://doi.org/10.3390/separations8010004
Chicago/Turabian StyleDadouch, Meriem, Yoann Ladner, and Catherine Perrin. 2021. "Analysis of Monoclonal Antibodies by Capillary Electrophoresis: Sample Preparation, Separation, and Detection" Separations 8, no. 1: 4. https://doi.org/10.3390/separations8010004
APA StyleDadouch, M., Ladner, Y., & Perrin, C. (2021). Analysis of Monoclonal Antibodies by Capillary Electrophoresis: Sample Preparation, Separation, and Detection. Separations, 8(1), 4. https://doi.org/10.3390/separations8010004