
Expanding the Knowledge Related to Flavors and Fragrances by Means of Three-Dimensional Preparative Gas Chromatography and Molecular Spectroscopy

 , ,
, ,  ,
,  ,
,  and
and 
Abstract
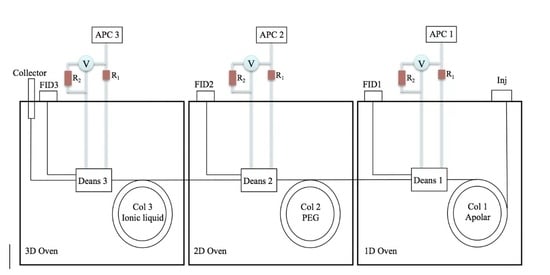
:
1. Introduction
2. Materials and Methods
2.1. Plant Material and Sample Preparation
2.2. GC-FID and GC-MS
2.3. Preparative Multidimensional GC
2.4. Nuclear Magnetic Resonance
2.5. Solid Phase GC-FTIR
3. Results
3.1. GC-MS and GC-FID Analyses
3.2. Preparative MDGC Analysis
3.3. Spectroscopic Data and Structural Elucidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hennia, A.; Nemmiche, S.; Dandlen, S.; Miguel, M.G. Myrtus communis essential oils: Insecticidal, antioxidant and antimicrobial activities: A review. J. Essent. Oil Res. 2019, 31, 487–545. [Google Scholar] [CrossRef]
- Migliore, J.; Baumel, A.; Juin, M.; Médail, F. From Mediterranean shores to central Saharan mountains: Key phylogeographical insights from the genus Myrtus. J. Biogeogr. 2012, 39, 942–956. [Google Scholar] [CrossRef]
- Sisay, M.; Gashaw, T. Ethnobotanical, Ethnopharmacological, and Phytochemical Studies of Myrtus communis Linn: A Popular Herb in Unani System of Medicine. Evid. Based Complement. Alternat. Med. 2017, 22, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Brada, M.; Tabti, N.; Boutoumi, H.; Wathelet, J.P.; Lognay, G. Composition of the essential oil of leaves and berries of Algerian myrtle (Myrtus communis L.). J. Essent. Oil Res. 2012, 24, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Bouzabata, A.; Castole, V.; Bighelli, A.; Abed, L.; Casanova, J.; Tomi, F. Chemical variability of Algerian Myrtus communis L. Chem. Biodivers. 2013, 10, 129–137. [Google Scholar] [CrossRef]
- Rahimmalek, M.; Mirzakhani, M.; Pirbalouti, A.G. Essential oil variation among 21 wild myrtle (Myrtus communis L.) populations collected from different geographical regions in Iran. Ind. Crops Prod. 2013, 51, 328–333. [Google Scholar] [CrossRef]
- Bazzali, O.; Tomi, F.; Casanova, J.; Bighelli, A. Occurrence of C8-C10 esters in Mediterranean Myrtus communis L. leaf essential oil. Flav. Fragr. J. 2012, 27, 335–340. [Google Scholar] [CrossRef]
- Hennia, A.; Miguel, M.G.; Brada, M.; Nemmiche, S.; Figueiredo, A.C. Composition, chemical variability and effect of distillation time on leaf and fruits essential oils of Myrtus communis from north western Algeria. J. Essent. Oil Res. 2016, 28, 146–156. [Google Scholar] [CrossRef]
- Pereira, P.; Cebola, M.; Oliveira, M.C.; Bernardo-Gil, M.G. Supercritical fluid extraction vs conventional extraction of myrtle leaves and berries: Comparison of antioxidant activity and identification of bioactive compounds. J. Supercrit. Fluids 2016, 113, 1–9. [Google Scholar] [CrossRef]
- Berka-Zougali, B.; Ferhat, M.A.; Hassani, A.; Chemat, F.; Allaf, K.S. Comparative Study of Essential Oils Extracted from Algerian Myrtus communis L. Leaves Using Microwaves and Hydrodistillation. Int. J. Mol. Sci. 2012, 13, 4673–4695. [Google Scholar] [CrossRef] [Green Version]
- Fadil, M.; Farah, A.; Ihssane, B.; Haloui, T.; Lebrazi, S.; Rachiq, S. Intrapopulation variability of Myrtus communis L. growing in Morocco: Chemometric investigation and antibacterial activity. J. Appl. Res. Med. Aromat. Plants 2017, 7, 35–40. [Google Scholar] [CrossRef]
- Wannes, W.A.; Mhamdi, B.; Sriti, J.; Marzouk, B. Changes in Essential Oil Composition of Tunisian Myrtus communis var. italica L. During Its Vegetative Cycle. J. Essent. Oil Res. 2010, 22, 13–18. [Google Scholar] [CrossRef]
- David, F.; Devos, C.; Sandra, P. Classical two-dimensional GC combined with mass spectrometry. LCGC Eur. 2006, 19, 602–616. [Google Scholar] [CrossRef]
- Amaral, M.S.S.; Marriott, P. The Blossoming of Technology for the Analysis of Complex Aroma Bouquets—A Review on Flavour and Odorant Multidimensional and Comprehensive Gas Chromatography Applications. Molecules 2019, 24, 2080. [Google Scholar] [CrossRef] [Green Version]
- Mondello, L.; Casilli, A.; Tranchida, P.Q.; Sciarrone, D.; Dugo, P.; Dugo, G. Analysis of Allergens in Fragrances using Multiple Heart-cut Multidimensional Gas Chromatography–Mass Spectrometry. LCGC Eur. 2008, 21, 130–137. [Google Scholar]
- Dong, G.; Bai, X.; Aimila, A.; Aisa, H.A.; Maiwulanjiang, M. Study on Lavender Essential Oil Chemical Compositions by GC-MS and Improved pGC. Molecules 2020, 25, 3166. [Google Scholar] [CrossRef]
- Li, Y.; Dong, G.; Bai, X.; Aimila, A.; Bai, X.; Maiwulanjiang, M.; Aisa, H.A. Separation and qualitative study of Ruta graveolens L. essential oil components by prep-GC. GC-QTOF-MS and NMR. Nat. Prod. Res. 2020, 35, 4202–4205. [Google Scholar] [CrossRef]
- Rahmani, R.; Andersson, F.; Andersson, M.N.; Yuvaraj, J.K.; Anderbrant, O.; Hedenström, E. Identification of sesquisabinene B in carrot (Daucus carota L.) leaves as a compound electrophysiologically active to the carrot psyllid (Trioza apicalis Förster). Chemoecology 2019, 29, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Eyres, G.T.; Urban, S.; Morrison, P.D.; Marriott, P.J. Application of microscale-preparative multidimensional gas chromatography with nuclear magnetic resonance spectroscopy for identification of pure methylnaphthalenes from crude oils. J. Chromatogr. A 2008, 1215, 168–176. [Google Scholar] [CrossRef]
- Eyres, G.T.; Urban, S.; Morrison, P.D.; Dufour, J.P.; Marriott, P.J. Method for Small-Molecule Discovery Based on Microscale-Preparative Multidimensional Gas Chromatography Isolation with Nuclear Magnetic Resonance Spectroscopy. Anal. Chem. 2008, 80, 6293–6299. [Google Scholar] [CrossRef]
- Sciarrone, D.; Pantò, S.; Rotondo, A.; Tedone, L.; Tranchida, P.Q.; Dugo, P.; Mondello, L. Rapid collection and identification of a novel component from Clausena lansium Skeels leaves by means of three-dimensional preparative gas chromatography and nuclear magnetic resonance/infrared/mass spectrometric analysis. Anal. Chim. Acta 2013, 785, 119–125. [Google Scholar] [CrossRef]
- Sciarrone, D.; Pantò, S.; Tranchida, P.Q.; Dugo, P.; Mondello, L. Rapid Isolation of High Solute Amounts Using an Online Four-Dimensional Preparative System: Normal Phase-Liquid Chromatography Coupled to Methyl Siloxane–Ionic Liquid–Wax Phase Gas Chromatography. Anal. Chem. 2014, 86, 4295–4301. [Google Scholar] [CrossRef]
- Sciarrone, D.; Pantò, S.; Donato, P.; Mondello, L. Improving the productivity of a multidimensional chromatographic preparative system by collecting pure chemicals after each of three chromatographic dimensions. J. Chromatogr. A 2016, 1475, 80–85. [Google Scholar] [CrossRef]
- Sciarrone, D.; Giuffrida, D.; Rotondo, A.; Micalizzi, G.; Zoccali, M.; Pantò, S.; Donato, P.; Rodrigues-das-Dores, R.G.; Mondello, L. Quali-quantitative characterization of the volatile constituents in Cordia verbenacea D.C. essential oil exploiting advanced chromatographic approaches and nuclear magnetic resonance analysis. J. Chromatogr. A 2017, 1524, 246–253. [Google Scholar] [CrossRef]
- Sciarrone, D.; Schepis, A.; De Grazia, G.; Rotondo, A.; Alibrando, F.; Cipriano, R.R.; Bizzo, H.; Deschamps, C.; Sidisky, L.M.; Mondello, L. Collection and identification of an unknown component from Eugenia uniflora essential oil exploiting a multidimensional preparative three-GC system employing apolar, mid-polar and ionic liquid stationary phases. Faraday Discuss. 2019, 218, 101–114. [Google Scholar] [CrossRef]
- Sciarrone, D.; Panto, S.; Ragonese, C.; Tranchida, P.Q.; Dugo, P.; Mondello, L. Increasing the Isolated Quantities and Purities of Volatile Compounds by Using a Triple Deans-Switch Multidimensional Preparative Gas Chromatographic System with an Apolar-Wax-Ionic Liquid Stationary-Phase Combination. Anal. Chem. 2012, 84, 7092–7098. [Google Scholar] [CrossRef]
- Rotondo, A.; Ettari, R.; Zappalà, M.; De Micheli, C.; Rotondo, E. NMR characterization and conformational analysis of a potent papain-family cathepsin L-like cysteine protease inhibitor with different behaviour in polar and apolar media. J. Mol. Struct. 2014, 1076, 337–343. [Google Scholar] [CrossRef]
- Rotondo, A.; Ettari, R.; Grasso, S.; Zappalà, M. NMR conformational analysis in solution of a potent class of cysteine proteases inhibitors. Struct. Chem. 2015, 26, 943–950. [Google Scholar] [CrossRef]
- Farah, A.; Afifi, A.; Fechtal, M.; Chhen, A.; Satrani, B.; Talbi, M.; Chaouch, A. Fractional distillation effect on the chemical composition of Moroccan myrtle (Myrtus communis L.) essential oils. Flavour Fragr. J. 2006, 21, 351–354. [Google Scholar] [CrossRef]
- Weyerstahl, P.; Marschall, H.; Rustaiyan, A. Constituents of the Essential Oil of Myrtus communis L. from Iran. Flavour Fragr. J. 1994, 9, 333. [Google Scholar] [CrossRef]
- Paolini, J.; Leandri, J.C.; Desjobert, J.-M.; Barboni, T.; Costa, J. Comparison of liquid–liquid extraction with headspace methods for the characterization of volatile fractions of commercial hydrolats from typically Mediterranean species. J. Chromatogr. A 2008, 1193, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Salerno, T.M.G.; Donato, P.; Frison, G.; Zamengo, L.; Mondello, L. Gas Chromatography-Fourier Transform Infrared Spectroscopy for Unambiguous Determination of Illicit Drugs: A Proof of Concept. Front. Chem. 2020, 8, 624. [Google Scholar] [CrossRef] [PubMed]
- Frison, G.; Zancanaro, F.; Frasson, S.; Quadretti, L.; Agnati, M.; Vlassich, F.; Gagliardi, G.; Salerno, T.M.G.; Donato, P.; Mondello, L. Analytical Characterization of 3-MeO-PCP and 3-MMC in Seized Products and Biosamples: The Role of LC-HRAM-Orbitrap-MS and Solid Deposition GC-FTIR. Front. Chem. 2021, 8, 618339. [Google Scholar] [CrossRef]
- Salerno, T.M.G.; Coppolino, C.; Donato, P.; Mondello, L. The online coupling of liquid chromatography to Fourier transform infrared spectroscopy using a solute-deposition interface: A proof of concept. Anal. Bioanal. Chem. 2022, 414, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.; Marto, J.; Duarte, A.; Gonçalves, L.M.; Pinto, P.; Figueiredo, A.C.; Ribeiro, H.M. Characterization of Portuguese Thymbra capitata, Thymus caespititius and Myrtus communis essential oils in topical formulations. Flavour Fragr. J. 2017, 32, 392–402. [Google Scholar] [CrossRef]
- Liu, H.; Huo, L.; Yang, B.; Yuan, Y.; Zhang, W.; Xu, Z.; Qiu, S.; Tan, H. Biomimetic-Inspired Syntheses of Myrtucommuacetalone and Myrtucommulone. J. Org. Lett. 2017, 19, 4786–4789. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Compounds | LRItheor | LRIexp | Essential Oil | LRIexp | Hydrosol |
|---|---|---|---|---|---|---|
| 1 | Isobutyl isobutyrate | 913 | 912 | 0.64 | 912 | 0.15 |
| 2 | α-Thujene | 927 | 925 | 0.19 | - | - |
| 3 | α-Pinene | 933 | 934 | 14.61 | 933 | 1.75 |
| 4 | Camphene | 953 | 948 | 0.08 | - | - |
| 5 | β-Pinene | 978 | 977 | 0.51 | 977 | 0.05 |
| 6 | 6-methyl-Hept-5-en-2-one | 986 | - | - | 984 | 0.07 |
| 7 | trans-5-Isopropenyl-2-methyl-2-vinyl-tetrahydrofuran | 989 | 990 | 0.13 | 990 | 0.14 |
| 8 | Myrcene | 991 | 989 | 0.04 | - | - |
| 9 | isobutyl 2-methyl Butyrate | 1002 | 1002 | 0.57 | 1002 | 0.13 |
| 10 | cis-dehydro-Linalool oxide | 1006 | 1006 | 0.19 | 1006 | 0.12 |
| 11 | δ-3-Carene | 1009 | 1009 | 0.08 | - | - |
| 12 | Isopentyl isobutyrate | 1014 | 1015 | 0.23 | 1014 | 0.04 |
| 13 | p-Cymene | 1025 | 1026 | 0.68 | 1025 | 0.08 |
| 14 | Limonene | 1030 | 1029 | 3.54 | 1029 | 1.03 |
| 15 | Eucalyptol | 1032 | 1035 | 27.25 | 1033 | 22.46 |
| 16 | Benzyl alcohol | 1040 | - | - | 1036 | 0.91 |
| 17 | trans-β-Ocimene | 1046 | 1046 | 0.08 | - | - |
| 18 | γ-Terpinene | 1058 | 1058 | 0.09 | - | - |
| 19 | cis-Linalool oxide | 1069 | - | - | 1070 | 0.71 |
| 20 | m-Cresol | 1073 | - | - | 1078 | 0.04 |
| 21 | trans-Linalool oxide | 1086 | - | - | 1086 | 0.60 |
| 22 | Terpinolene | 1086 | 1086 | 0.11 | - | - |
| 23 | p-Cymenene | 1093 | 1092 | 0.07 | - | - |
| 24 | Linalool | 1101 | 1101 | 4.94 | 1101 | 4.81 |
| 25 | 3-methylbutyl-2-methyl-Butyrate | 1104 | 1103 | 1.24 | - | - |
| 26 | trans-p-Mentha-2,8-dien-1-ol | 1122 | - | - | 1123 | 0.04 |
| 27 | Fenchyl alcohol | 1123 | 1120 | 0.05 | 1120 | 0.09 |
| 28 | α-Campholenal | 1125 | - | - | 1127 | 0.03 |
| 29 | Limona ketone | 1131 | - | - | 1132 | 0.04 |
| 30 | cis-Limonene oxide | 1134 | 1134 | 0.05 | - | - |
| 31 | cis-p-Mentha-2,8-dien-1-ol | 1138 | - | - | 1137 | 0.07 |
| 32 | Nopinone | 1139 | - | - | 1140 | 0.06 |
| 33 | trans-Pinocarveol | 1141 | 1142 | 0.34 | 1142 | 0.84 |
| 34 | trans-Verbenol | 1145 | 1148 | 0.09 | - | - |
| 35 | cis-β-Terpineol | 1149 | - | - | 1150 | 0.15 |
| 36 | Camphene hydrate | 1156 | 1156 | 0.08 | ||
| 37 | Menthone | 1158 | 1157 | 0.04 | - | - |
| 38 | Pinocarvone | 1164 | 1164 | 0.06 | 1163 | 0.04 |
| 39 | δ-Terpineol | 1170 | 1171 | 0.24 | 1171 | 0.98 |
| 40 | Borneol | 1173 | 1173 | 0.06 | 1173 | 0.15 |
| 41 | trans-Linalool oxide (pyranoid) | 1174 | 1176 | - | 1176 | 0.15 |
| 42 | Terpinen-4-ol | 1184 | 1181 | 0.58 | 1181 | 1.58 |
| 43 | α-Terpineol | 1195 | 1198 | 7.14 | 1200 | 22.74 |
| 44 | Myrtenol | 1202 | 1199 | 2.14 | 1201 | 3.85 |
| 45 | Verbenone | 1208 | - | - | 1210 | 0.13 |
| 46 | Fenchyl acetate | 1219 | 1218 | 0.04 | - | - |
| 47 | trans-Carveol | 1223 | 1222 | 0.10 | 1222 | 0.67 |
| 48 | cis-p-Mentha-1(7),8-dien-2-ol | 1230 | 1230 | 0.06 | 1232 | 0.43 |
| 49 | cis-Carveol | 1232 | - | - | 1235 | 0.11 |
| 50 | Pulegone | 1241 | 1240 | 0.63 | 1240 | 2.02 |
| 51 | Linalyl acetate | 1250 | 1250 | 0.20 | - | - |
| 52 | Geraniol | 1255 | 1253 | 0.68 | 1253 | 2.59 |
| 53 | Geranial | 1268 | - | - | 1269 | 0.05 |
| 54 | trans-Pinocarvyl acetate | 1296 | 1296 | 0.65 | 1296 | 0.18 |
| 55 | Myrtenyl acetate | 1324 | 1327 | 15.96 | 1327 | 5.16 |
| 56 | α-Terpenyl acetate | 1349 | 1348 | 1.39 | 1347 | 0.43 |
| 57 | cis-Geranyl acetate | 1361 | 1360 | 0.15 | - | - |
| 58 | α-Copaene | 1375 | 1376 | 0.10 | - | - |
| 59 | trans-Geranyl acetate | 1380 | 1379 | 4.22 | 1378 | 1.22 |
| 60 | trans-Myrtanol acetate | 1387 | 1385 | 0.32 | - | - |
| 61 | β-Elemene | 1390 | 1390 | 0.12 | - | - |
| 62 | Methyl eugenol | 1403 | 1403 | 3.33 | 1405 | 8.50 |
| 63 | β-Caryophyllene | 1424 | 1420 | 1.11 | 1419 | 0.17 |
| 64 | Perillyl acetate | 1435 | 1436 | 0.06 | - | - |
| 65 | α-Humulene | 1454 | 1456 | 0.47 | - | - |
| 66 | β-Santalene | 1459 | 1459 | 0.05 | - | - |
| 67 | Myrtenyl isobutyrate | 1463 | 1463 | 0.34 | - | - |
| 68 | β-Selinene | 1492 | 1489 | 0.24 | - | - |
| 69 | α-Selinene | 1501 | 1497 | 0.14 | - | - |
| 70 | Geranyl isobutyrate | 1507 | 1508 | 0.42 | - | - |
| 71 | Unknown | - | 1515 | 0.78 | 1515 | 7.29 |
| 72 | Elemicin | 1548 | - | - | 1550 | 0.53 |
| 73 | Caryophyllene oxide | 1587 | 1583 | 0.65 | 1583 | 0.26 |
| 74 | Geranyl 2-methylbutyrate | 1596 | 1597 | 0.19 | - | - |
| 75 | Humulene epoxide II | 1613 | 1612 | 0.24 | 1611 | 0.07 |
| TOT | 98.60 | 93.79 |
| 13C Label | 13C Shift | Type | 1H Label | 1H Shift | 13C Calc Shift | 1H Calc Shift | 13C Count | 1H Count | 1H HMBC | 13C HMBC |
|---|---|---|---|---|---|---|---|---|---|---|
| C 2 | 90.8 | C | 87.93 | 8, 8′, 3 | ||||||
| C 3 | 39.6 | CH2 | H 3 | 2.749 | 37.73 | 2.706 | 1 | 2 | 8, 8′ | 8, 8′, 2, 3′, 7′ |
| C 3′ | 110 | C | 112.96 | 3 | ||||||
| C 4 | 195 | C | 193.51 | 9′, 9 | ||||||
| C 5 | 55 | C | 55.74 | 9′, 9 | ||||||
| C 6 | 214 | C | 210.81 | 9′, 9, 10′, 10 | ||||||
| C 7 | 45.3 | C | 45.06 | 10′, 10 | ||||||
| C 7′ | 177 | C | 171.22 | 10′, 10, 3 | ||||||
| C 8 | 28.1 | CH3 | H 8 | 1.482 | 28.39 | 1.411 | 2 | 6 | 3 | 3, 2 |
| C 9 | 24.6 | CH3 | H 9 | 1.351 | 21.25 | 1.335 | 2 | 6 | 5, 4, 6 | |
| C 10 | 24.2 | CH3 | H 10 | 1.391 | 23.29 | 1.348 | 2 | 6 | 7, 7′, 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Grazia, G.; Cucinotta, L.; Rotondo, A.; Donato, P.; Mondello, L.; Sciarrone, D. Expanding the Knowledge Related to Flavors and Fragrances by Means of Three-Dimensional Preparative Gas Chromatography and Molecular Spectroscopy. Separations 2022, 9, 202. https://doi.org/10.3390/separations9080202
De Grazia G, Cucinotta L, Rotondo A, Donato P, Mondello L, Sciarrone D. Expanding the Knowledge Related to Flavors and Fragrances by Means of Three-Dimensional Preparative Gas Chromatography and Molecular Spectroscopy. Separations. 2022; 9(8):202. https://doi.org/10.3390/separations9080202
Chicago/Turabian StyleDe Grazia, Gemma, Lorenzo Cucinotta, Archimede Rotondo, Paola Donato, Luigi Mondello, and Danilo Sciarrone. 2022. "Expanding the Knowledge Related to Flavors and Fragrances by Means of Three-Dimensional Preparative Gas Chromatography and Molecular Spectroscopy" Separations 9, no. 8: 202. https://doi.org/10.3390/separations9080202
APA StyleDe Grazia, G., Cucinotta, L., Rotondo, A., Donato, P., Mondello, L., & Sciarrone, D. (2022). Expanding the Knowledge Related to Flavors and Fragrances by Means of Three-Dimensional Preparative Gas Chromatography and Molecular Spectroscopy. Separations, 9(8), 202. https://doi.org/10.3390/separations9080202