
Probable Reasons for Neuron Copper Deficiency in the Brain of Patients with Alzheimer’s Disease: The Complex Role of Amyloid



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Copper Physiology Focusing on the Brain
3. Connections between Copper and Aβ
4. Copper’s Importance in the Hippocampus
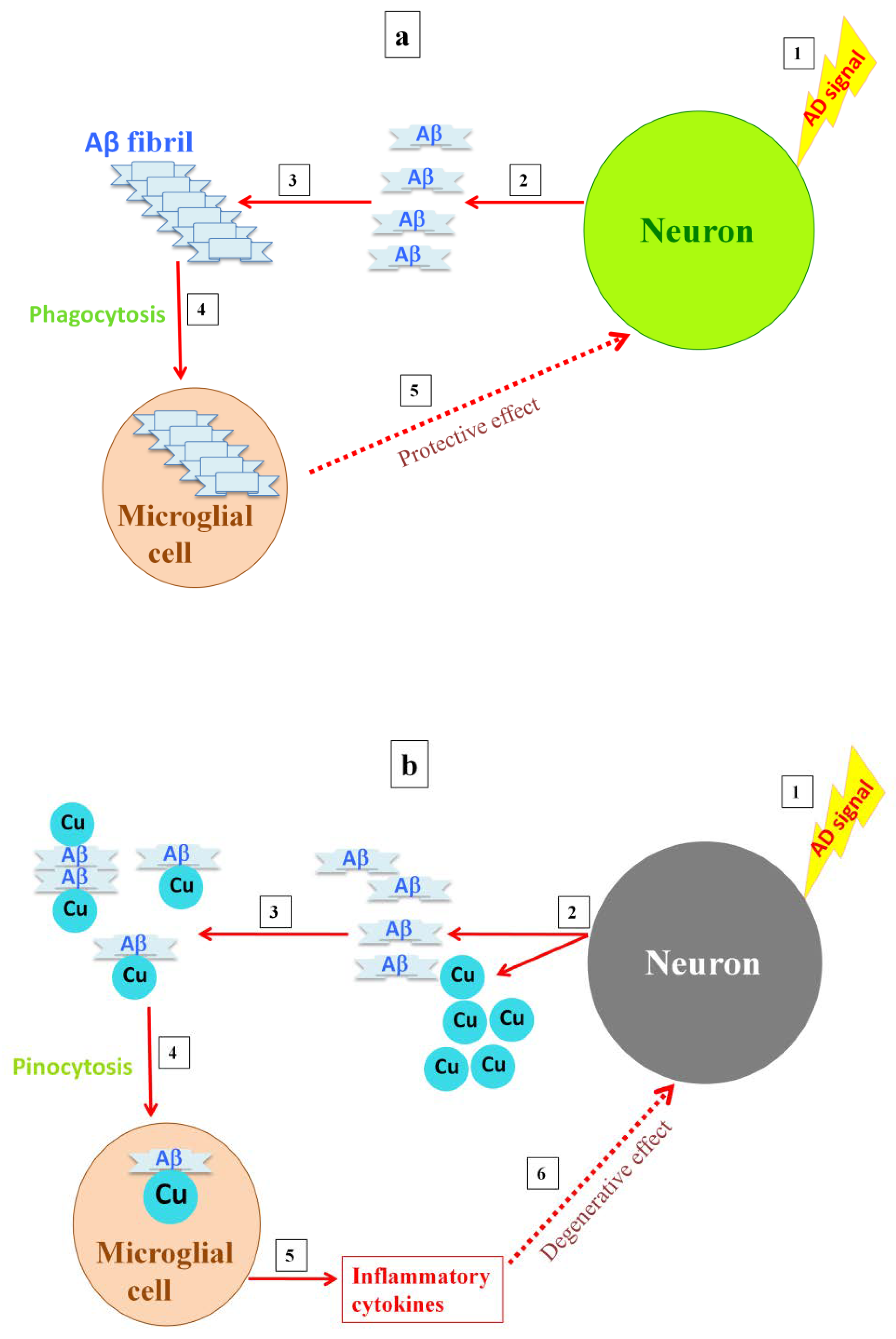
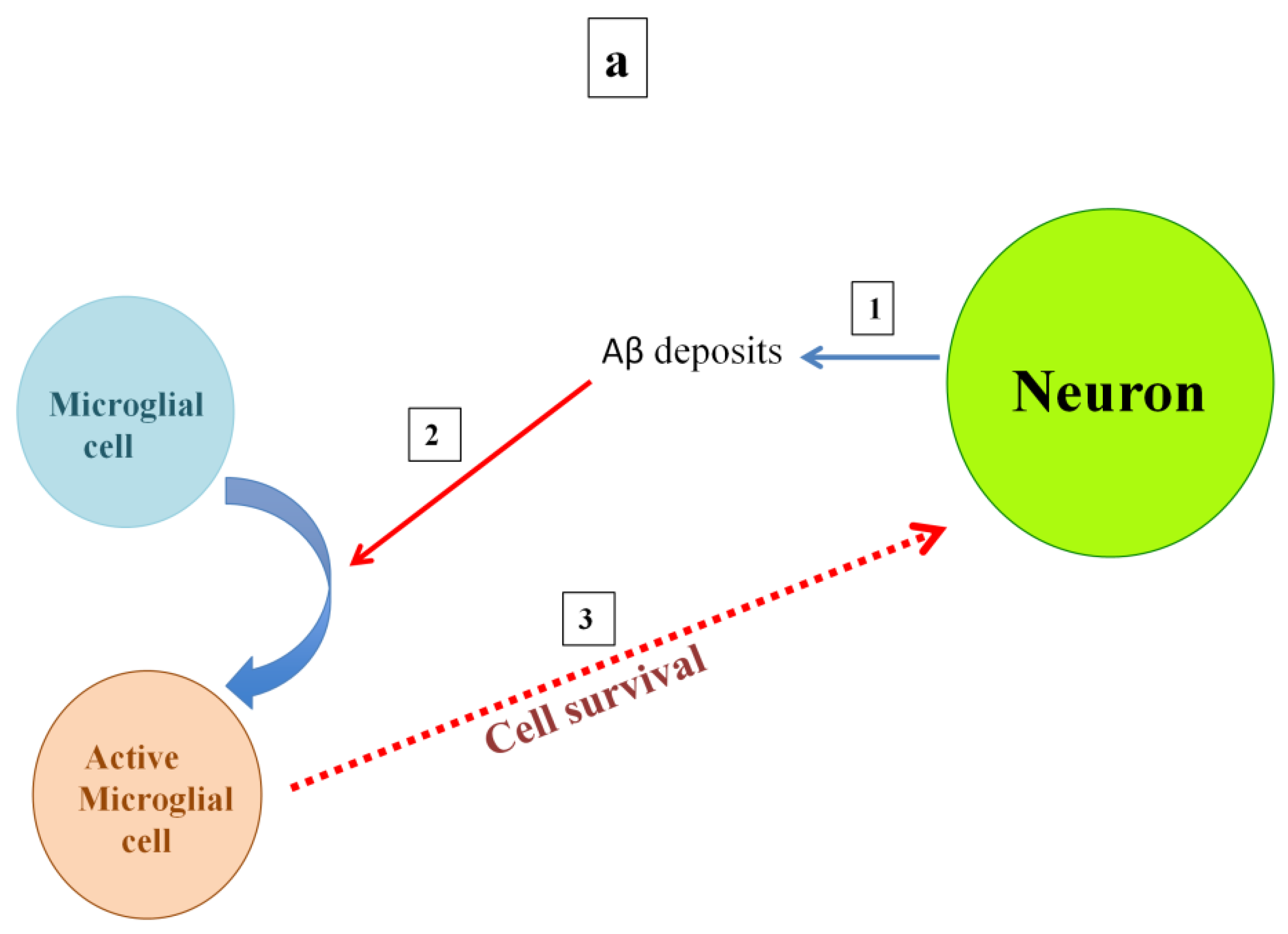
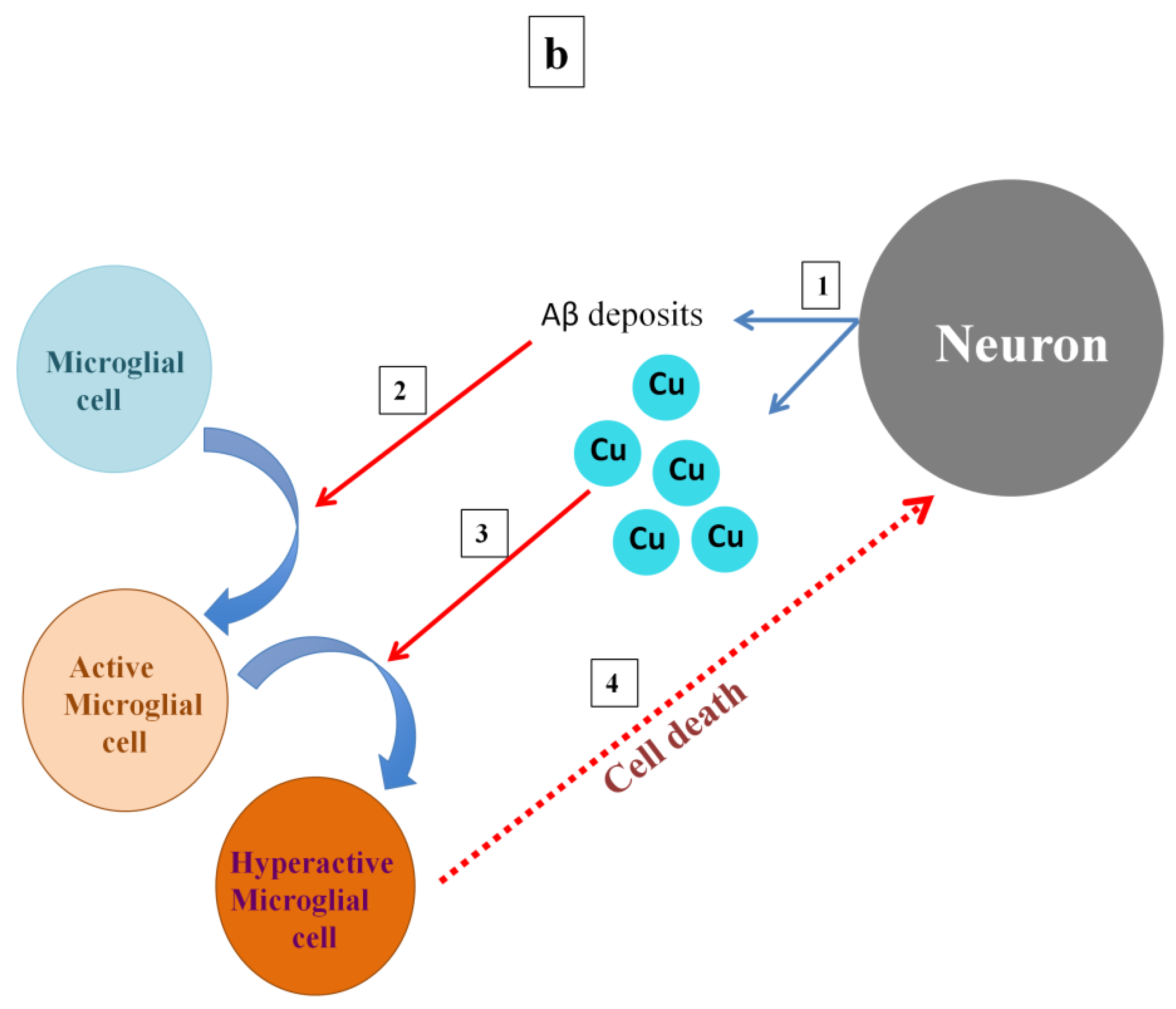
5. Roles of Microglia in Both Healthy and AD Brains
6. Interplay of Copper, Aβ, and Microglia Activation in Mediating NMDA Receptor-Induced Excitotoxicity
7. Insights and Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Australia, D.; Baker, S.; Banerjee, S. Alzheimer’s Disease International World Alzheimer Report 2019: Attitudes to Dementia. Available online: https://www.alzint.org/resource/world-alzheimer-report-2019/ (accessed on 20 September 2019).
- Liu, P.P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P. Ten Challenges of the Amyloid Hypothesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef]
- De Benedictis, C.A.; Vilella, A.; Grabrucker, A.M. The Role of Trace Metals in Alzheimer’s Disease. In Alzheimer’s Disease; Codon Publications: Brisbane, Australia, 2019; pp. 85–106. ISBN 9780646809687. [Google Scholar]
- Bagheri, S.; Saboury, A.A. What role do metals play in Alzheimer’s disease? J. Iran. Chem. Soc. 2021, 18, 2199–2213. [Google Scholar] [CrossRef]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Kepp, K.P.; Squitti, R. Copper imbalance in Alzheimer’s disease: Convergence of the chemistry and the clinic. Coord. Chem. Rev. 2019, 397, 168–187. [Google Scholar] [CrossRef]
- Squitti, R.; Simonelli, I.; Cassetta, E.; Ventriglia, M.; Lupoi, D.; Rongioletti, M.; Siotto, M. Patients with increased non-ceruloplasmin copper appear a distinct sub-group of alzheimer’s disease: A neuroimaging study. Curr. Alzheimer Res. 2017, 14, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Ventriglia, M.; Gennarelli, M.; Colabufo, N.A.; El Idrissi, I.G.; Bucossi, S.; Mariani, S.; Rongioletti, M.; Zanetti, O.; Congiu, C.; et al. Non-Ceruloplasmin Copper Distincts Subtypes in Alzheimer’s Disease: A Genetic Study of ATP7B Frequency. Mol. Neurobiol. 2017, 54, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Schrag, M.; Mueller, C.; Oyoyo, U.; Smith, M.A.; Kirsch, W.M. Iron, zinc and copper in the Alzheimer’s disease brain: A quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 2011, 94, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squitti, R.; Ghidoni, R.; Simonelli, I.; Ivanova, I.D.; Colabufo, N.A.; Zuin, M.; Benussi, L.; Binetti, G.; Cassetta, E.; Rongioletti, M.; et al. Copper dyshomeostasis in Wilson disease and Alzheimer’s disease as shown by serum and urine copper indicators. J. Trace Elem. Med. Biol. 2018, 45, 181–188. [Google Scholar] [CrossRef]
- Rembach, A.; Hare, D.J.; Lind, M.; Fowler, C.J.; Cherny, R.A.; Mclean, C.A.; Bush, A.I.; Masters, C.L.; Roberts, B.R. Decreased copper in Alzheimer’s disease brain is predominantly in the soluble extractable fraction. Int. J. Alzheimer’s Dis. 2013, 2013, 623241. [Google Scholar] [CrossRef]
- Akatsu, H.; Hori, A.; Yamamoto, T.; Yoshida, M.; Mimuro, M.; Hashizume, Y.; Tooyama, I.; Yezdimer, E.M. Transition metal abnormalities in progressive dementias. Biometals 2012, 25, 337–350. [Google Scholar] [CrossRef]
- Magaki, S.; Raghavan, R.; Mueller, C.; Oberg, K.C.; Vinters, H.V.; Kirsch, W.M. Iron, copper, and iron regulatory protein 2 in Alzheimer’s disease and related dementias. Neurosci. Lett. 2007, 418, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; McHarg, S.; Kureishy, N.; Hollywood, K.A.; Waldvogel, H.J.; Liu, H.; Zhang, S.; et al. Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer’s disease: Metabolic basis for dementia. Sci. Rep. 2016, 6, 27524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deibel, M.A.; Ehmann, W.D.; Markesbery, W.R. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. J. Neurol. Sci. 1996, 143, 137–142. [Google Scholar] [CrossRef]
- Scholefield, M.; Church, S.J.; Xu, J.; Patassini, S.; Roncaroli, F.; Hooper, N.M.; Unwin, R.D.; Cooper, G.J.S. Widespread Decreases in Cerebral Copper Are Common to Parkinson’s Disease Dementia and Alzheimer’s Disease Dementia. Front. Aging Neurosci. 2021, 13, 81. [Google Scholar] [CrossRef]
- James, S.A.; Volitakis, I.; Adlard, P.A.; Duce, J.A.; Masters, C.L.; Cherny, R.A.; Bush, A.I. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 298–302. [Google Scholar] [CrossRef]
- Giacoppo, S.; Galuppo, M.; Calabrò, R.S.; D’Aleo, G.; Marra, A.; Sessa, E.; Bua, D.G.; Potortì, A.G.; Dugo, G.; Bramanti, P.; et al. Heavy Metals and Neurodegenerative Diseases: An Observational Study. Biol. Trace Elem. Res. 2014, 161, 151–160. [Google Scholar] [CrossRef]
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of copper in the onset of Alzheimer’s disease compared to other metals. Front. Aging Neurosci. 2018, 9, 446. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.L.; Liu, X.Z.; Shen, P.; Zheng, Y.G.; Lan, X.R.; Lu, C.B.; Wang, J.Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Malosio, M.L.; Tecchio, F.; Squitti, R. Molecular mechanisms underlying copper function and toxicity in neurons and their possible therapeutic exploitation for Alzheimer’s disease. Aging Clin. Exp. Res. 2021, 33, 2027–2030. [Google Scholar] [CrossRef]
- Nose, Y.; Kim, B.E.; Thiele, D.J. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006, 4, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaler, S.G. ATP7A-related copper transport diseasesg-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Lutsenko, S. Human copper homeostasis: A network of interconnected pathways. Curr. Opin. Chem. Biol. 2010, 14, 211–217. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Lavado, L.K.; Zhang, M.; Patel, K.; Khan, S.; Patel, U.K. Biometals as Potential Predictors of the Neurodegenerative Decline in Alzheimer’s Disease. Cureus 2019, 11, e5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- German, N.; Doyscher, D.; Rensing, C. Bacterial killing in macrophages and amoeba: Do they all use a brass dagger? Future Microbiol. 2013, 8, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Lüthje, F.L.; Qin, Y.; McDevitt, S.F.; Lutay, N.; Hobman, J.L.; Asiani, K.; Soncini, F.C.; German, N.; Zhang, S.; et al. Survival in amoeba—a major selection pressure on the presence of bacterial copper and zinc resistance determinants? Identification of a “copper pathogenicity island”. Appl. Microbiol. Biotechnol. 2015, 99, 5817–5824. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Lüthje, F.; Rønn, R.; German, N.A.; Li, X.; Huang, F.; Kisaka, J.; Huffman, D.; Alwathnani, H.A.; Zhu, Y.G.; et al. A role for copper in protozoan grazing–two billion years selecting for bacterial copper resistance. Mol. Microbiol. 2016, 102, 628–641. [Google Scholar] [CrossRef]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, I.F.; Mercer, J.F.B.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.S.; Zheng, W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009, 1248, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Chen, J.; Zheng, W. Relative contribution of CTR1 and DMT1 in copper transport by the blood-CSF barrier: Implication in manganese-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2012, 260, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W. Neurotoxicology of the brain barrier system: New implications. J. Toxicol.-Clin. Toxicol. 2001, 39, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Zhang, J.; Xu, Y.; Shen, X.; Song, H.; Jing, J.; Luo, W.; Zheng, W.; Chen, J. Involvement of CTR1 and ATP7A in lead (Pb)-induced copper (Cu) accumulation in choroidal epithelial cells. Toxicol. Lett. 2014, 225, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Zhang, Y.; Jiang, W.; Monnot, A.D.; Bates, C.A.; Zheng, W. Regulation of copper transport crossing brain barrier systems by CU-ATPases: Effect of manganese exposure. Toxicol. Sci. 2014, 139, 432–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrowolska, J.; Dehnhardt, M.; Matusch, A.; Zoriy, M.; Palomero-Gallagher, N.; Koscielniak, P.; Zilles, K.; Becker, J.S. Quantitative imaging of zinc, copper and lead in three distinct regions of the human brain by laser ablation inductively coupled plasma mass spectrometry. Talanta 2008, 74, 717–723. [Google Scholar] [CrossRef]
- Davies, K.M.; Hare, D.J.; Cottam, V.; Chen, N.; Hilgers, L.; Halliday, G.; Mercer, J.F.B.; Double, K.L. Localization of copper and copper transporters in the human brain. Metallomics 2013, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, E.; Salazar, E.; Villasmil, J.J.; Villalobos, R.; Gonzalez, M.; Davila, J.O. Copper distribution in the normal human brain. Neurochem. Res. 1984, 9, 1543–1548. [Google Scholar] [CrossRef]
- Wang, L.M.; Becker, J.S.; Wu, Q.; Oliveira, M.F.; Bozza, F.A.; Schwager, A.L.; Hoffman, J.M.; Morton, K.A. Bioimaging of copper alterations in the aging mouse brain by autoradiography, laser ablation inductively coupled plasma mass spectrometry and immunohistochemistry. Metallomics 2010, 2, 348–353. [Google Scholar] [CrossRef]
- Serpa, R.F.B.; De Jesus, E.F.O.; Anjos, M.J.; De Oliveira, L.F.; Marins, L.A.; Do Carmo, M.G.T.; Corrêa, J.D.; Rocha, M.S.; Lopes, R.T.; Martinez, A.M.B. Topographic trace-elemental analysis in the brain of wistar rats by X-ray microfluorescence with synchrotron radiation. Anal. Sci. 2008, 24, 839–842. [Google Scholar] [CrossRef] [Green Version]
- Tarohda, T.; Yamamoto, M.; Amamo, R. Regional distribution of manganese, iron, copper, and zinc in the rat brain during development. Anal. Bioanal. Chem. 2004, 380, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.; Santos, A.; Pinto, N.R.; Mendes, R.; Magalhães, T.; Almeida, A. Anatomical Region Differences and Age-Related Changes in Copper, Zinc, and Manganese Levels in the Human Brain. Biol. Trace Elem. Res. 2014, 161, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Nasaruddin, M.B.; Carey, M.; Holscher, C.; McGuinness, B.; Kehoe, P.G.; Love, S.; Passmore, P.; Elliott, C.T.; Meharg, A.A.; et al. Age-associated changes of brain copper, iron, and zinc in Alzheimer’s disease and dementia with Lewy bodies. J. Alzheimer’s Dis. 2014, 42, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef] [Green Version]
- Southon, A.; Greenough, M.A.; Ganio, G.; Bush, A.I.; Burke, R.; Camakaris, J. Presenilin Promotes Dietary Copper Uptake. PLoS ONE 2013, 8, e62811. [Google Scholar] [CrossRef] [Green Version]
- Tümer, Z.; Møller, L.B. Menkes disease. Eur. J. Hum. Genet. 2010, 18, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Montes, S.; Rivera-Mancia, S.; Diaz-Ruiz, A.; Tristan-Lopez, L.; Rios, C. Copper and Copper Proteins in Parkinson’s Disease. Oxid. Med. Cell. Longev. 2014, 2014, 147251. [Google Scholar] [CrossRef] [Green Version]
- Deloncle, R.; Guillard, O. Is brain copper deficiency in Alzheimer’s, lewy body, and Creutzfeldt Jakob diseases the common key for a free radical mechanism and oxidative stress-induced damage? J. Alzheimer’s Dis. 2014, 43, 1149–1156. [Google Scholar] [CrossRef]
- Scholefield, M.; Unwin, R.D.; Cooper, G.J.S. Shared perturbations in the metallome and metabolome of Alzheimer’s, Parkinson’s, Huntington’s, and dementia with Lewy bodies: A systematic review. Ageing Res. Rev. 2020, 63, 101152. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, D.J.; Bartnikas, T.B.; Gitlin, J.D. The role of copper in neurodegenerative disease. Neurobiol. Dis. 1999, 6, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s Disease Amyloid-β Binds Copper and Zinc to Generate an Allosterically Ordered Membrane-penetrating Structure Containing Superoxide Dismutase-like Subunits. J. Biol. Chem. 2001, 276, 20466–20473. [Google Scholar] [CrossRef] [Green Version]
- Ciccotosto, G.D.; Tew, D.J.; Drew, S.C.; Smith, D.G.; Johanssen, T.; Lal, V.; Lau, T.L.; Perez, K.; Curtain, C.C.; Wade, J.D.; et al. Stereospecific interactions are necessary for Alzheimer disease amyloid-β toxicity. Neurobiol. Aging 2011, 32, 235–248. [Google Scholar] [CrossRef]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep. 2016, 6, 28781. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Azimova, R.; Kagan, B.L.; Nussinov, R.; Lal, R. Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6538–6543. [Google Scholar] [CrossRef] [Green Version]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Retamal, C.; Cuitiño, L.; Caruano-Yzermans, A.; Shin, J.E.; Van Kerkhof, P.; Marzolo, M.P.; Bu, G. Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J. Biol. Chem. 2008, 283, 11501–11508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, E.; Dolev, I.; Fogel, H.; Ciccotosto, G.D.; Ruff, E.; Slutsky, I. Amyloid-Β as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 2009, 12, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Macreadie, I. Copper transport and Alzheimer’s disease. Eur. Biophys. J. 2008, 37, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Atwood, C.S.; Moir, R.D.; Huang, X.; Scarpa, R.C.; Bacarra, N.M.E.; Romano, D.M.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Dramatic aggregation of alzheimer by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 1998, 273, 12817–12826. [Google Scholar] [CrossRef] [Green Version]
- Hesse, L.; Beher, D.; Masters, C.L.; Multhaup, G. The βA4 amyloid precursor protein binding to copper. FEBS Lett. 1994, 349, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Treiber, C.; Simons, A.; Strauss, M.; Hafner, M.; Cappai, R.; Bayer, T.A.; Multhaup, G. Clioquinol mediates copper uptake and counteracts copper efflux activities of the amyloid precursor protein of Alzheimer’s disease. J. Biol. Chem. 2004, 279, 51958–51964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, C.J.; Cappai, R.; Volitakis, I.; Cherny, R.A.; White, A.R.; Beyreuther, K.; Masters, C.L.; Bush, A.I.; Li, Q.X. Overexpression of Alzheimer’s disease amyloid-β opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 2002, 277, 44670–44676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarell, C.J.; Syme, C.D.; Rigby, S.E.J.; Viles, J.H. Copper(II) binding to amyloid-β fibrils of Alzheimer’s disease reveals a picomolar affinity: Stoichiometry and coordination geometry are independent of Aβ oligomeric form. Biochemistry 2009, 48, 4388–4402. [Google Scholar] [CrossRef]
- Barritt, J.D.; Viles, J.H. Truncated amyloid-β(11-40/42) from Alzheimer disease binds Cu2+ with a femtomolar affinity and influences fiber assembly. J. Biol. Chem. 2015, 290, 27791–27802. [Google Scholar] [CrossRef] [Green Version]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of Copper Interactions with Alzheimer Amyloid β Peptides. J. Neurochem. 2008, 75, 1219–1233. [Google Scholar] [CrossRef]
- Wezynfeld, N.E.; Stefaniak, E.; Stachucy, K.; Drozd, A.; Płonka, D.; Drew, S.C.; Krężel, A.; Bal, W. Resistance of Cu(Aβ4-16) to Copper Capture by Metallothionein-3 Supports a Function for the Aβ4-42 Peptide as a Synaptic Cu II Scavenger. Angew. Chemie Int. Ed. 2016, 55, 8235–8238. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Rothman, S.M.; Olney, J.W. Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann. Neurol. 1986, 19, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar]
- Tymianski, M.; Charlton, M.P.; Carlen, P.L.; Tator, C.H. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J. Neurosci. 1993, 13, 2085–2104. [Google Scholar] [CrossRef]
- Choi, D.W. Ionic dependence of glutamate neurotoxicity. J. Neurosci. 1987, 7, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.L.; Landau, S.M.; Bell, R.K.; Jagust, W.J. Hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline. Elife 2017, 6, e22978. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, W.; Mormino, E.C.; Schultz, A.P.; Wigman, S.; Ward, A.M.; Larvie, M.; Amariglio, R.E.; Marshall, G.A.; Rentz, D.M.; Johnson, K.A.; et al. Amyloid-β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 2015, 138, 1023–1035. [Google Scholar] [CrossRef] [Green Version]
- Kodis, E.J.; Choi, S.; Swanson, E.; Ferreira, G.; Bloom, G.S. N-methyl-d-aspartate receptor–mediated calcium influx connects amyloid-β oligomers to ectopic neuronal cell cycle reentry in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 1302–1312. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Dhikav, V.; Anand, K. Potential predictors of hippocampal atrophy in alzheimers disease. Drugs Aging 2011, 28, 1–11. [Google Scholar] [CrossRef]
- Gosche, K.M.; Mortimer, J.A.; Smith, C.D.; Markesbery, W.R.; Snowdon, D.A. Hippocampal volume as an index of Alzheimer neuropathology: Findings from the Nun study. Neurology 2002, 58, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Scahill, R.I.; Schott, J.M.; Stevens, J.M.; Rossor, M.N.; Fox, N.C. Mapping the evolution of regional atrophy in Alzheimer’s disease: Unbiased analysis of fluid-registered serial MRI. Proc. Natl. Acad. Sci. USA 2002, 99, 4703–4707. [Google Scholar] [CrossRef] [Green Version]
- Hartter, D.E.; Barnea, A. Evidence for release of copper in the brain: Depolarization-induced release of newly taken-up 67copper. Synapse 1988, 2, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Hopt, A.; Korte, S.; Fink, H.; Panne, U.; Niessner, R.; Jahn, R.; Kretzschmar, H.; Herms, J. Methods for studying synaptosomal copper release. J. Neurosci. Methods 2003, 128, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Kovács, I.; Hajós, F.; Kálmán, M.; Simonyi, M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar] [CrossRef]
- Schlief, M.L.; Craig, A.M.; Gitlin, J.D. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 2005, 25, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.C.; Kinghorn, K.J.; Woodling, N.S. Shifting equilibriums in Alzheimer’s disease: The complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural Regen. Res. 2020, 15, 1208–1219. [Google Scholar]
- You, H.; Tsutsui, S.; Hameed, S.; Kannanayakal, T.J.; Chen, L.; Xia, P.; Engbers, J.D.T.; Lipton, S.A.; Stys, P.K.; Zamponi, G.W. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, L.; Meneghetti, E.; Pastore, B.; Benetti, F.; Legname, G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal. 2015, 22, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Tsiroulnikov, K.; Chobert, J.-M.; Haertlé, T. Copper-dependent degradation of recombinant ovine prion protein. FEBS J. 2006, 273, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Schlief, M.L.; West, T.; Craig, A.M.; Holtzman, D.M.; Gitlin, J.D. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc. Natl. Acad. Sci. USA 2006, 103, 14919–14924. [Google Scholar] [CrossRef] [Green Version]
- Dodani, S.C.; Firl, A.; Chan, J.; Nam, C.I.; Aron, A.T.; Onak, C.S.; Ramos-Torres, K.M.; Paek, J.; Webster, C.M.; Feller, M.B.; et al. Copper is an endogenous modulator of neural circuit spontaneous activity. Proc. Natl. Acad. Sci. USA 2014, 111, 16280–16285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filiano, A.J.; Gadani, S.P.; Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015, 1617, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicastro, N.; Mak, E.; Williams, G.B.; Surendranathan, A.; Bevan-Jones, W.R.; Passamonti, L.; Vàzquez Rodrìguez, P.; Su, L.; Arnold, R.; Fryer, T.D.; et al. Correlation of microglial activation with white matter changes in dementia with Lewy bodies. NeuroImage Clin. 2020, 25, 102200. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Ninan, S.; Atkinson, R.; Fan, Z.; Brooks, D.J.; Edison, P. Does microglial activation influence hippocampal volume and neuronal function in Alzheimer’s disease and Parkinson’s disease dementia? J. Alzheimer’s Dis. 2016, 51, 1275–1289. [Google Scholar] [CrossRef]
- Nicastro, N.; Surendranathan, A.; Mak, E.; Rowe, J.B.; O’Brien, J.T. 11C-PK11195 PET imaging and white matter changes in Parkinson’s disease dementia. Ann. Clin. Transl. Neurol. 2019, 6, 2133–2136. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Mohammad, D.; Trépanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Dani, M.; Wood, M.; Fan, Z.; Calsolaro, V.; Atkinson, R.; Edginton, T.; Hinz, R.; Brooks, D.J.; Edison, P. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 2019, 92, 1331–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Petris, M.J.; Mercer, J.F.B.; Culvenor, J.G.; Lockhart, P.; Gleeson, P.A.; Camakaris, J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: A novel mechanism of regulated trafficking. EMBO J. 1996, 15, 6084–6095. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Heiny, M.E.; Suzuki, M.; Gitlin, J.D. Biochemical characterization and intracellular localization of the Menkes disease protein. Proc. Natl. Acad. Sci. USA 1996, 93, 14030–14035. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; White, C.; Lee, J.; Peterson, T.S.; Bush, A.I.; Sun, G.Y.; Weisman, G.A.; Petris, M.J. Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J. Neurochem. 2010, 114, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Achard, M.E.S.; Stafford, S.L.; Bokil, N.J.; Chartres, J.; Bernhardt, P.V.; Schembri, M.A.; Sweet, M.J.; Mcewan, A.G. Copper redistribution in murine macrophages in response to Salmonella infection. Biochem. J. 2012, 444, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.; Maser, J.; Lai, B.; Cai, Z.; Barry, C.E.; Höner zu Bentrup, K.; Russell, D.G.; Bermudez, L.E. Elemental Analysis of Mycobacterium avium -, Mycobacterium tuberculosis -, and Mycobacterium smegmatis -Containing Phagosomes Indicates Pathogen-Induced Microenvironments within the Host Cell’s Endosomal System. J. Immunol. 2005, 174, 1491–1500. [Google Scholar] [CrossRef] [Green Version]
- Festa, R.A.; Thiele, D.J. Copper at the Front Line of the Host-Pathogen Battle. PLoS Pathog. 2012, 8, e1002887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.; Lee, J.; Kambe, T.; Fritsche, K.; Petris, M.J. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 2009, 284, 33949–33956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Ouyang, Z.; Tan, Y.; Wu, H.; Liu, Y. Activating macrophages for enhanced osteogenic and bactericidal performance by Cu ion release from micro/nano-topographical coating on a titanium substrate. Acta Biomater. 2019, 100, 415–426. [Google Scholar] [CrossRef]
- Lim, S.L.; Rodriguez-Ortiz, C.J.; Hsu, H.W.; Wu, J.; Zumkehr, J.; Kilian, J.; Vidal, J.; Ayata, P.; Kitazawa, M. Chronic copper exposure directs microglia towards degenerative expression signatures in wild-type and J20 mouse model of Alzheimer’s disease. J. Trace Elem. Med. Biol. 2020, 62, 126578. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and Their Roles in Health and Disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Long, T.; Pan, Q.; Zhang, S.; Zhang, Y.; Zhang, D.; Qin, G.; Chen, L.; Zhou, J. Microglial NLRP3 inflammasome activation mediates IL-1β release and contributes to central sensitization in a recurrent nitroglycerin-induced migraine model. J. Neuroinflamm. 2019, 16, 78. [Google Scholar] [CrossRef] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Ojala, J.; Suuronen, T.; Kaarniranta, K.; Kauppinen, A. Amyloid-β oligomers set fire to inflammasomes and induce Alzheimer’s pathology: Alzheimer Review Series. J. Cell. Mol. Med. 2008, 12, 2255–2262. [Google Scholar] [CrossRef]
- Deigendesch, N.; Zychlinsky, A.; Meissner, F. Copper Regulates the Canonical NLRP3 Inflammasome. J. Immunol. 2018, 200, 1607–1617. [Google Scholar] [CrossRef]
- Dong, J.; Wang, X.; Xu, C.; Gao, M.; Wang, S.; Zhang, J.; Tong, H.; Wang, L.; Han, Y.; Cheng, N.; et al. Inhibiting NLRP3 inflammasome activation prevents copper-induced neuropathology in a murine model of Wilson’s disease. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Tago, H.; McGeer, E.G. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci. Lett. 1987, 79, 195–200. [Google Scholar] [CrossRef]
- Haga, S.; Akai, K.; Ishii, T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol. 1989, 77, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.W. Amyloid-β protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706. [Google Scholar] [CrossRef] [Green Version]
- Jekabsone, A.; Mander, P.K.; Tickler, A.; Sharpe, M.; Brown, G.C. Fibrillar beta-amyloid peptide Aβ1-40 activates microglial proliferation via stimulating TNF-α release and H2O2 derived from NADPH oxidase: A cell culture study. J. Neuroinflamm. 2006, 3, 24. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tan, M.-S.; Jiang, T.; Tan, L. Microglia in Alzheimer’s Disease. Biomed Res. Int. 2014, 2014, 437483. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Ajit, D.; Peterson, T.S.; Wang, Y.; Camden, J.M.; Wood, W.G.; Sun, G.Y.; Erb, L.; Petris, M.; Weisman, G.A. Nucleotides released from Aβ 1-42-treated microglial cells increase cell migration and Aβ 1-42 uptake through P2Y 2 receptor activation. J. Neurochem. 2012, 121, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Q.; Chen, C.; Dou, Y.; Wu, H.-J.; Liu, Y.-J.; Lou, H.-F.; Zhang, J.-M.; Li, X.-M.; Wang, H.; Duan, S. P2Y4 Receptor-Mediated Pinocytosis Contributes to Amyloid Beta-Induced Self-Uptake by Microglia. Mol. Cell. Biol. 2013, 33, 4282–4293. [Google Scholar] [CrossRef] [Green Version]
- Swanson, J.A. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 2008, 9, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.D.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble aβ through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Khoury, J.; Hickman, S.E.; Thomas, C.A.; Cao, L.; Silverstein, S.C.; Loike, J.D. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 1996, 382, 716–719. [Google Scholar] [CrossRef]
- Pan, X.D.; Zhu, Y.G.; Lin, N.; Zhang, J.; Ye, Q.Y.; Huang, H.P.; Chen, X.C. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- James, S.A.; Churches, Q.I.; De Jonge, M.D.; Birchall, I.E.; Streltsov, V.; McColl, G.; Adlard, P.A.; Hare, D.J. Iron, Copper, and Zinc Concentration in Aβ Plaques in the APP/PS1 Mouse Model of Alzheimer’s Disease Correlates with Metal Levels in the Surrounding Neuropil. ACS Chem. Neurosci. 2017, 8, 629–637. [Google Scholar] [CrossRef]
- Barnham, K.J. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease -amyloid. FASEB J. 2004, 18, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Sarell, C.J.; Wilkinson, S.R.; Viles, J.H. Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-β from Alzheimer disease. J. Biol. Chem. 2010, 285, 41533–41540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.P.; Ciccotosto, G.D.; Tew, D.J.; Fodero-Tavoletti, M.T.; Johanssen, T.; Masters, C.L.; Barnham, K.J.; Cappai, R. Concentration dependent Cu2+ induced aggregation and dityrosine formation of the Alzheimer’s disease amyloid-βpeptide. Biochemistry 2007, 46, 2881–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of Metal Ions in the Self-assembly of the Alzheimer ’s Amyloid β Peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar] [CrossRef]
- Tõugu, V.; Karafin, A.; Zovo, K.; Chung, R.S.; Howells, C.; West, A.K.; Palumaa, P. Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-β (1-42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 2009, 110, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Gong, P.; Hu, Z.; Qiu, Y.; Cui, Y.; Gao, X.; Chen, H.; Li, J. Cu(II) enhances the effect of Alzheimer’s amyloid-β peptide on microglial activation. J. Neuroinflamm. 2015, 12, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazawa, M.; Hsu, H.W.; Medeiros, R. Copper exposure perturbs brain inflammatory responses and impairs clearance of amyloid-beta. Toxicol. Sci. 2016, 152, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Guan, H.; Yang, Y.; Luo, S.; Hou, L.; Chen, H.; Li, J. Cu(II) disrupts autophagy-mediated lysosomal degradation of oligomeric Aβ in microglia via mTOR-TFEB pathway. Toxicol. Appl. Pharmacol. 2020, 401, 115090. [Google Scholar] [CrossRef]
- Mayes, J.; Tinker-Mill, C.; Kolosov, O.; Zhang, H.; Tabner, B.J.; Allsop, D. β-Amyloid fibrils in alzheimer disease are not inert when bound to copper ions but can degrade hydrogen peroxide and generate reactive oxygen species. J. Biol. Chem. 2014, 289, 12052–12062. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.L.; Sun, Y.X.; Jiang, Z.F. Cu(II) potentiation of Alzheimer Aβ1-40 cytotoxicity and transition on its secondary structure. Acta Biochim. Biophys. Sin. 2006, 38, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Church, S.J.; Patassini, S.; Begley, P.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.M.; Unwin, R.D.; Cooper, G.J.S. Evidence for widespread, severe brain copper deficiency in Alzheimer’s dementia. Metallomics 2017, 9, 1106–1119. [Google Scholar] [CrossRef] [Green Version]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Abad-Rodriguez, J.; Ledesma, M.D.; Craessaerts, K.; Perga, S.; Medina, M.; Delacourte, A.; Dingwall, C.; De Strooper, B.; Dotti, C.G. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 2004, 167, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Chung, C.Y.S.; Liu, P.; Craciun, L.; Nishikawa, Y.; Bruemmer, K.J.; Hamachi, I.; Saijo, K.; Miller, E.W.; Chang, C.J. Activity-Based Sensing with a Metal-Directed Acyl Imidazole Strategy Reveals Cell Type-Dependent Pools of Labile Brain Copper. J. Am. Chem. Soc. 2020, 142, 14993–15003. [Google Scholar] [CrossRef] [PubMed]
- He, W.; James Kang, Y. Ischemia-induced copper loss and suppression of angiogenesis in the pathogenesis of myocardial infarction. Cardiovasc. Toxicol. 2013, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Berenshtein, E.; Mayer, B.; Goldberg, C.; Kitrossky, N.; Chevion, M. Patterns of mobilization of copper and iron following myocardial ischemia: Possible predictive criteria for tissue injury. J. Mol. Cell. Cardiol. 1997, 29, 3025–3034. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, C.; Xiao, Y.; Wang, T.; James Kang, Y. The loss of copper is associated with the increase in copper metabolism MURR domain 1 in ischemic hearts of mice. Exp. Biol. Med. 2018, 243, 780–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Perez, A.; Llansola, M.; Cauli, O.; Felipo, V. Modulation of NMDA receptors in the cerebellum. II. Signaling pathways and physiological modulators regulating NMDA receptor function. Cerebellum 2005, 4, 162–170. [Google Scholar] [CrossRef]
- Swanger, S.A.; Vance, K.M.; Pare, J.F.; Sotty, F.; Fog, K.; Smith, Y.; Traynelis, S.F. NMDA receptors containing the GluN2D subunit control neuronal function in the subthalamic nucleus. J. Neurosci. 2015, 35, 15971–15983. [Google Scholar] [CrossRef] [Green Version]
- Mullasseril, P.; Hansen, K.B.; Vance, K.M.; Ogden, K.K.; Yuan, H.; Kurtkaya, N.L.; Santangelo, R.; Orr, A.G.; Le, P.; Vellano, K.M.; et al. A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat. Commun. 2010, 1, 90. [Google Scholar] [CrossRef] [Green Version]
- Llansola, M.; Sanchez-Perez, A.; Cauli, O.; Felipo, V. Modulation of NMDA receptors in the cerebellum. 1. Properties of the NMDA receptor that modulate its function. Cerebellum 2005, 4, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Larner, A.J. The Cerebellum in alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 1997, 8, 203–209. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bagheri, S.; Saboury, A.A.; Haertlé, T.; Rongioletti, M.; Saso, L. Probable Reasons for Neuron Copper Deficiency in the Brain of Patients with Alzheimer’s Disease: The Complex Role of Amyloid. Inorganics 2022, 10, 6. https://doi.org/10.3390/inorganics10010006
Bagheri S, Saboury AA, Haertlé T, Rongioletti M, Saso L. Probable Reasons for Neuron Copper Deficiency in the Brain of Patients with Alzheimer’s Disease: The Complex Role of Amyloid. Inorganics. 2022; 10(1):6. https://doi.org/10.3390/inorganics10010006
Chicago/Turabian StyleBagheri, Soghra, Ali A. Saboury, Thomas Haertlé, Mauro Rongioletti, and Luciano Saso. 2022. "Probable Reasons for Neuron Copper Deficiency in the Brain of Patients with Alzheimer’s Disease: The Complex Role of Amyloid" Inorganics 10, no. 1: 6. https://doi.org/10.3390/inorganics10010006
APA StyleBagheri, S., Saboury, A. A., Haertlé, T., Rongioletti, M., & Saso, L. (2022). Probable Reasons for Neuron Copper Deficiency in the Brain of Patients with Alzheimer’s Disease: The Complex Role of Amyloid. Inorganics, 10(1), 6. https://doi.org/10.3390/inorganics10010006