
Digold Phosphinine Complexes Are Stable with a Bis(Phosphinine) Ligand but Not with a 2-Phosphinophosphinine



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
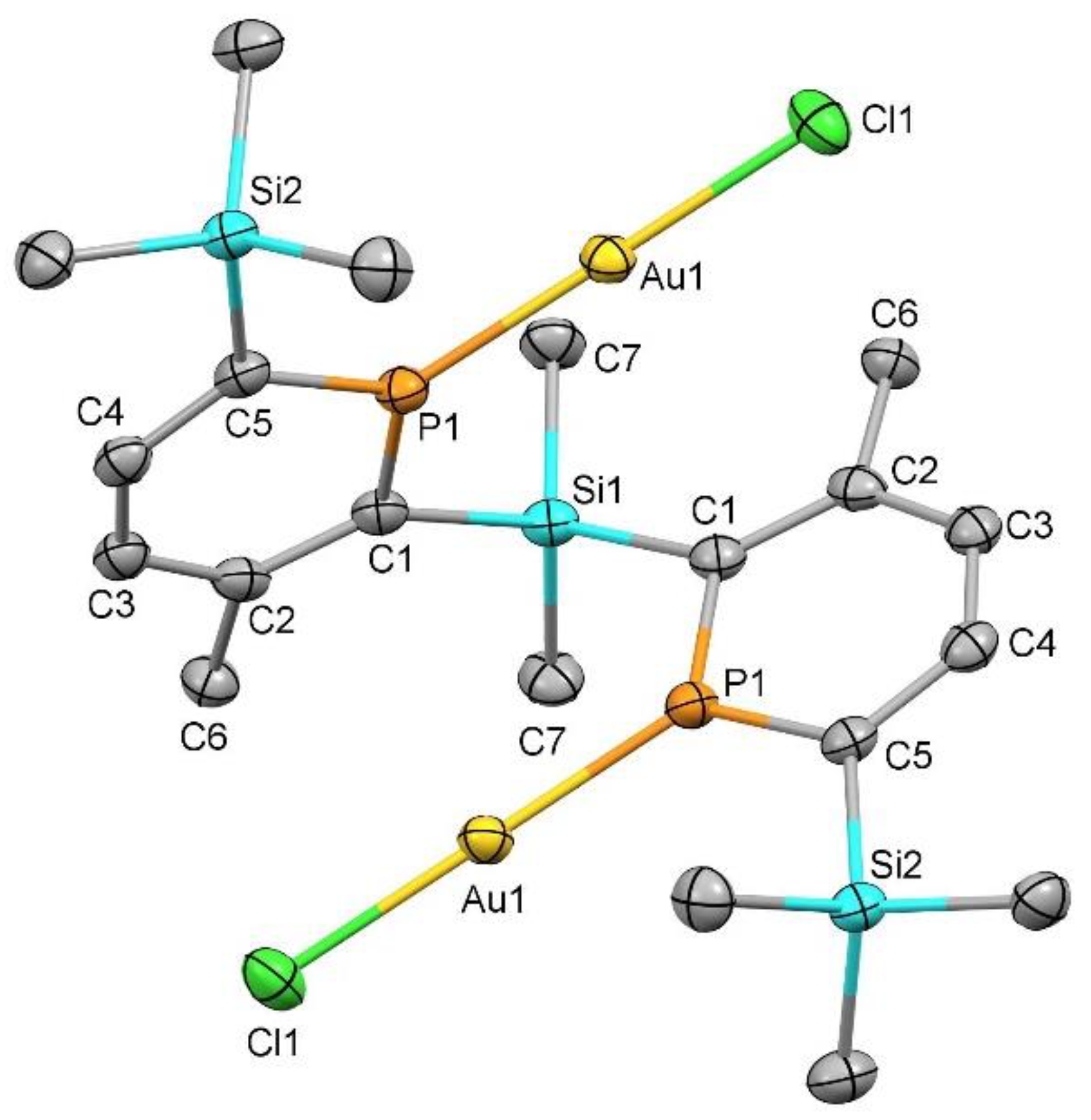
2. Results
3. Discussion
4. Materials and Methods
4.1. Attempted preparation of 20
4.2. Preparation of 21
4.3. Reactions of 22 with [AuCl(L)]
4.4. Data for 23:
4.5. Data for 24:
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Märkl, G. 2,4,6-Triphenylphosphabenzene. Angew. Chem. Int. Ed. 1966, 5, 846–847. [Google Scholar] [CrossRef]
- Ashe, A.J. Phosphabenzene and arsabenzene. J. Am. Chem. Soc. 1971, 93, 3293–3295. [Google Scholar] [CrossRef]
- Ashe, A.J. The Route to Phosphabenzene and Beyond. Eur. J. Inorg. Chem. 2016, 2016, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.; Vogt, D. Phosphinines as ligands in homogeneous catalysis: Recent developments, concepts and perspectives. Dalton Trans. 2007, 47, 5505–5523. [Google Scholar] [CrossRef]
- Müller, C.; Vogt, D. Recent developments in the chemistry of donor-functionalized phosphinines. C. R. Chim. 2010, 13, 1127–1143. [Google Scholar] [CrossRef]
- Müller, C.; Vogt, D. Phosphinine-Based Ligands in Homogeneous Catalysis: State of the Art and Future Perspectives. In Phosphorus Compounds. Catalysis by Metal Complexes; Peruzzini, M., Gonsalvi, L., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 37. [Google Scholar]
- Müller, C. Phosphinine Ligands. In Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 287–307. [Google Scholar]
- Müller, C.; Broeckx, L.E.E.; de Krom, I.; Weemers, J.J.M. Developments in the Coordination Chemistry of Phosphinines. Eur. J. Inorg. Chem. 2013, 2013, 187–202. [Google Scholar] [CrossRef]
- Müller, C.; Sklorz, J.A.W.; de Krom, I.; Loibl, A.; Habicht, M.; Bruce, M.; Pfeifer, G.; Wiecko, J. Recent Developments in the Chemistry of Pyridyl-functionalized, Low-coordinate Phosphorus Heterocycles. Chem. Lett. 2014, 43, 1390–1404. [Google Scholar] [CrossRef] [Green Version]
- Coles, N.T.; Sofie Abels, A.; Leitl, J.; Wolf, R.; Grützmacher, H.; Müller, C. Phosphinine-based ligands: Recent developments in coordination chemistry and applications. Coord. Chem. Rev. 2021, 433, 213729. [Google Scholar] [CrossRef]
- Weber, L. Phosphorus Heterocycles: From Laboratory Curiosities to Ligands in Highly Efficient Catalysts. Angew. Chem. Int. Ed. Engl. 2002, 41, 563–572. [Google Scholar] [CrossRef]
- Kostova, I.; Bentham Science, P. Gold Coordination Complexes as Anticancer Agents. Anti-Cancer Agents Med. Chem. 2006, 6, 19–32. [Google Scholar] [CrossRef]
- Best, S.L.; Sadler, P.J. Gold drugs: Mechanism of action and toxicity. Gold Bull. 1996, 29, 87–93. [Google Scholar] [CrossRef]
- Hutchings, G.J. Catalysis by gold. Catal. Today 2005, 100, 55–61. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Rudolph, M. Gold catalysis in total synthesis. Chem. Soc. Rev. 2008, 37, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Gold-Catalyzed Organic Reactions. Chem. Rev. 2007, 107, 3180–3211. [Google Scholar] [CrossRef] [PubMed]
- Campeau, D.; León Rayo, D.F.; Mansour, A.; Muratov, K.; Gagosz, F. Gold-Catalyzed Reactions of Specially Activated Alkynes, Allenes, and Alkenes. Chem. Rev. 2021, 121, 8756–8867. [Google Scholar] [CrossRef]
- Dash, K.C.; Eberlein, J.; Schmidbaur, H. Gold(i) Halide Complexes of 2,4,6-Triphenyl-Phosphabenzene. Synth. React. Inorg. Met.-Org. Chem. 1973, 3, 375–380. [Google Scholar] [CrossRef]
- Stott, J.; Bruhn, C.; Siemeling, U. Crystal Structures of the Gold(I) Phosphinine Complexes [AuCl(C5H2P-2,6-Me2-4-Ph)] and [AuCl(C5H2P-2,4,6-Ph3)]. Z. Für Nat. B 2013, 68, 853–859. [Google Scholar] [CrossRef]
- Mézailles, N.; Ricard, L.; Mathey, F.; Le Floch, P. Synthesis, X-ray Crystal Structures and Reactivity Towards Alkynes of Gold(I)–Phosphinine Complexes. Eur. J. Inorg. Chem. 1999, 1999, 2233–2241. [Google Scholar] [CrossRef]
- Avarvari, N.; Mézailles, N.; Ricard, L.; Floch, P.L.; Mathey, F. Silacalix-[n]-phosphaarenes: Macrocyclic Ligands Based on Dicoordinate Phosphorus Centers. Science 1998, 280, 1587–1589. [Google Scholar] [CrossRef]
- Avarvari, N.; Le Floch, P.; Mathey, F. 1,3,2-Diazaphosphinines: New, Versatile Precursors of 1,2-Azaphosphinines and Polyfunctional Phosphinines. J. Am. Chem. Soc. 1996, 118, 11978–11979. [Google Scholar] [CrossRef]
- Avarvari, N.; Le Floch, P.; Ricard, L.; Mathey, F. 1,3,2-Diazaphosphinines and -Diazaarsinines as Precursors for Polyfunctional Phosphinines and Arsinines. Organometallics 1997, 16, 4089–4098. [Google Scholar] [CrossRef]
- Mézailles, N.; Avarvari, N.; Maigrot, N.; Ricard, L.; Mathey, F.; Le Floch, P.; Cataldo, L.; Berclaz, T.; Geoffroy, M. Gold(I) and Gold(0) Complexes of Phosphinine-Based Macrocycles. Angew. Chem. Int. Ed. Engl. 1999, 38, 3194–3197. [Google Scholar] [CrossRef]
- Moores, A.; Mézailles, N.; Maigrot, N.; Ricard, L.; Mathey, F.; Le Floch, P. Synthesis and Reactivity Towards Cationic Group 11 Metal Centers of an Extended Silacalix-[3]-phosphinine Macrocycle. Eur. J. Inorg. Chem. 2002, 2002, 2034–2039. [Google Scholar] [CrossRef]
- Doux, M.; Ricard, L.; Le Floch, P.; Mézailles, N. Group 11 metal complexes of SPS-based pincer ligands: Syntheses, X-ray structures and reactivity. Dalton Trans. 2004, 16, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Clendenning, S.B.; Hitchcock, P.B.; Lawless, G.A.; Nixon, J.F.; Tate, C.W. Differences in the η1-ligating properties of 2,4,6-tritertiarybutyl-phosphabenzene, PC5H2Bu3t and 2,4,6-tritertiarybutyl-1,3,5-triphosphabenzene, P3C3Bu3t. J. Organomet. Chem. 2010, 695, 717–720. [Google Scholar] [CrossRef]
- Falconer, R.L.; Russell, C.A. 1,3,5-Triphosphabenzenes: Synthesis, reactivity and theory. Coord. Chem. Rev. 2015, 297–298, 146–167. [Google Scholar] [CrossRef]
- Townsend, N.S.; Green, M.; Russell, C.A. Cationic Gold(I) Complexes of 2,4,6-Tri-tert-butyl-1,3,5-triphosphabenzene. Organometallics 2012, 31, 2543–2545. [Google Scholar] [CrossRef]
- Komath Mallissery, S.; Nieger, M.; Gudat, D. On the Surface Reactivity of Functionalized Phosphinines on Inorganic Supports. Z. Anorg. Allg. Chem. 2010, 636, 1354–1360. [Google Scholar] [CrossRef]
- Mao, Y.; Lim, K.M.H.; Li, Y.; Ganguly, R.; Mathey, F. The Original Coordination Chemistry of 2-Phosphaphenol with Copper(I) and Gold(I) Halides. Organometallics 2013, 32, 3562–3565. [Google Scholar] [CrossRef]
- Hou, Y.; Li, Z.; Li, Y.; Liu, P.; Su, C.-Y.; Puschmann, F.; Grützmacher, H. Making the unconventional μ2-P bridging binding mode more conventional in phosphinine complexes. Chem. Sci. 2019, 10, 3168–3180. [Google Scholar] [CrossRef]
- Moussa, J.; Chamoreau, L.M.; Amouri, H. Gold(I) complexes with a phosphinine ligand: Synthesis and structural characterization. RSC Adv. 2014, 4, 11539–11542. [Google Scholar] [CrossRef]
- Weemers, J.J.M.; van der Graaff, W.N.P.; Pidko, E.A.; Lutz, M.; Müller, C. Bulky Phosphinines: From a Molecular Design to an Application in Homogeneous Catalysis. Chem.-Eur. J. 2013, 19, 8991–9004. [Google Scholar] [CrossRef] [PubMed]
- Rigo, M.; Hettmanczyk, L.; Heutz, F.J.L.; Hohloch, S.; Lutz, M.; Sarkar, B.; Müller, C. Phosphinines versus mesoionic carbenes: A comparison of structurally related ligands in Au(i)-catalysis. Dalton Trans. 2017, 46, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Rigo, M.; Habraken, E.R.M.; Bhattacharyya, K.; Weber, M.; Ehlers, A.W.; Mézailles, N.; Slootweg, J.C.; Müller, C. Phosphinine-Based Ligands in Gold-Catalyzed Reactions. Chem.-Eur. J. 2019, 25, 8769–8779. [Google Scholar] [CrossRef]
- Yoshifuji, M. Chemistry of Several Sterically Bulky Molecules with P=P, P=C, and C≡P Bond. Molecules 2022, 27, 1557. [Google Scholar] [CrossRef] [PubMed]
- Freytag, M.; Ito, S.; Yoshifuji, M. Coordination Behavior of Sterically Protected Phosphaalkenes on the AuCl Moiety Leading to Catalytic 1,6-Enyne Cycloisomerization. Chem. -Asian J. 2006, 1, 693–700. [Google Scholar] [CrossRef]
- Franchino, A.; Montesinos-Magraner, M.; Echavarren, A.M. Silver-Free Catalysis with Gold(I) Chloride Complexes. Bull. Chem. Soc. Jpn. 2021, 94, 1099–1117. [Google Scholar] [CrossRef]
- Le Floch, P. Phosphaalkene, phospholyl and phosphinine ligands: New tools in coordination chemistry and catalysis. Coord. Chem. Rev. 2006, 250, 627–681. [Google Scholar] [CrossRef]
- Cleaves, P.A.; Mansell, S.M. Unexpected Multiple Coordination Modes in Silyl-Bridged Bis(phosphinine) Complexes. Organometallics 2019, 38, 1595–1605. [Google Scholar] [CrossRef]
- Cleaves, P.A.; Gourlay, B.; Newland, R.J.; Westgate, R.; Mansell, S.M. Reactivity Studies of Phosphinines: The Selenation of Diphenyl-Phosphine Substituents and Formation of a Chelating Bis(Phosphinine) Palladium(II) Complex. Inorganics 2022, 10, 17. [Google Scholar] [CrossRef]
- Newland, R.J.; Wyatt, M.F.; Wingad, R.L.; Mansell, S.M. A ruthenium(II) bis(phosphinophosphinine) complex as a precatalyst for transfer-hydrogenation and hydrogen-borrowing reactions. Dalton Trans. 2017, 46, 6172–6176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newland, R.J.; Delve, M.P.; Wingad, R.L.; Mansell, S.M. Two isomers of a bis(diphenylphosphino)-phosphinine, and the synthesis and reactivity of Ru arene/Cp* phosphinophosphinine complexes. N. J. Chem. 2018, 42, 19625–19636. [Google Scholar] [CrossRef] [Green Version]
- Mansell, S.M. Catalytic applications of small bite-angle diphosphorus ligands with single-atom linkers. Dalton Trans. 2017, 46, 15157–15174. [Google Scholar] [CrossRef]
- Newland, R.J.; Lynam, J.M.; Mansell, S.M. Small bite-angle 2-phosphinophosphinine ligands enable rhodium-catalysed hydroboration of carbonyls. Chem. Commun. 2018, 54, 5482–5485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newland, R.J.; Smith, A.; Smith, D.M.; Fey, N.; Hanton, M.J.; Mansell, S.M. Accessing Alkyl- and Alkenylcyclopentanes from Cr-Catalyzed Ethylene Oligomerization Using 2-Phosphinophosphinine Ligands. Organometallics 2018, 37, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Trodden, E.C.; Delve, M.P.; Luz, C.; Newland, R.J.; Andresen, J.M.; Mansell, S.M. A ruthenium cis-dihydride with 2-phosphinophosphinine ligands catalyses the acceptorless dehydrogenation of benzyl alcohol. Dalton Trans. 2021, 50, 13407–13411. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schier, A. Aurophilic interactions as a subject of current research: An up-date. Chem. Soc. Rev. 2012, 41, 370–412. [Google Scholar] [CrossRef]
- Caracelli, I.; Zukerman-Schpector, J.; Tiekink, E.R.T. Supra-molecular synthons based on gold…π(arene) interactions. Gold Bull. 2013, 46, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Habicht, M.H.; Wossidlo, F.; Bens, T.; Pidko, E.A.; Müller, C. 2-(Trimethylsilyl)-λ3-Phosphinine: Synthesis, Coordination Chemistry, and Reactivity. Chem.-Eur. J. 2018, 24, 944–952. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cleaves, P.A.; Gourlay, B.; Marseglia, M.; Ward, D.J.; Mansell, S.M. Digold Phosphinine Complexes Are Stable with a Bis(Phosphinine) Ligand but Not with a 2-Phosphinophosphinine. Inorganics 2022, 10, 203. https://doi.org/10.3390/inorganics10110203
Cleaves PA, Gourlay B, Marseglia M, Ward DJ, Mansell SM. Digold Phosphinine Complexes Are Stable with a Bis(Phosphinine) Ligand but Not with a 2-Phosphinophosphinine. Inorganics. 2022; 10(11):203. https://doi.org/10.3390/inorganics10110203
Chicago/Turabian StyleCleaves, Peter A., Ben Gourlay, Margot Marseglia, Daniel J. Ward, and Stephen M. Mansell. 2022. "Digold Phosphinine Complexes Are Stable with a Bis(Phosphinine) Ligand but Not with a 2-Phosphinophosphinine" Inorganics 10, no. 11: 203. https://doi.org/10.3390/inorganics10110203
APA StyleCleaves, P. A., Gourlay, B., Marseglia, M., Ward, D. J., & Mansell, S. M. (2022). Digold Phosphinine Complexes Are Stable with a Bis(Phosphinine) Ligand but Not with a 2-Phosphinophosphinine. Inorganics, 10(11), 203. https://doi.org/10.3390/inorganics10110203