
How to Protect ortho-Carborane from Decapitation—Practical Synthesis of 3,6-Dihalogen Derivatives 3,6-X2-1,2-C2B10H10 (X = Cl, Br, I)



Abstract
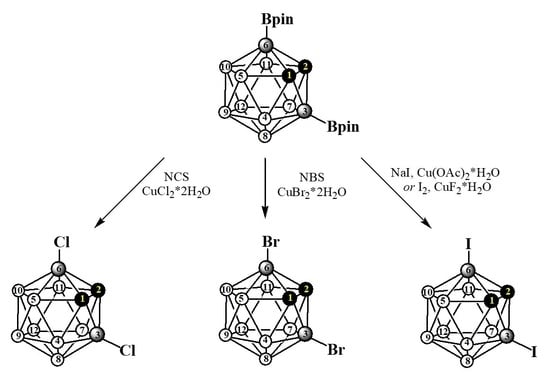
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of 3-Bpin-1,2-C2B10H11 (1) and 3,6-(Bpin)2-1,2-C2B10H10 (2)
3.3. General Procedure for the Synthesis of 3-Halogen-ortho-carboranes 3-X-1,2-C2B10H11 (X = Cl (3), Br(4))
3.4. Synthesis of 3-Iodo-ortho-Carborane 3-I-1,2-C2B10H11 (5)
3.5. Synthesis of 3-Acetoxy-ortho-Carborane 3-AcO-1,2-C2B10H11 (6)
3.6. General Procedure for the Synthesis of 3,6-Dihalogen-ortho-carboranes 3,6-X2-1,2-C2B10H10 (X = Cl (7), Br(8))
3.7. Synthesis of 3,6-Diiodo-ortho-Carborane 3,6-I2-1,2-C2B10H11 (9)
3.8. Synthesis of 3,6-Diacetoxy-ortho-Carborane 3,6-(AcO)2-1,2-C2B10H10 (10)
3.9. Synthesis of Cesium 3-chloro-7,8-Dicarba-Nido-Undecaborate Cs [3-Cl-7,8-C2B9H11] (11)
3.10. Single Crystal X-ray Diffraction Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiesboeck, R.A.; Hawthorne, M.F. Dicarbaundecaborane(13) and derivatives. J. Am. Chem. Soc. 1964, 86, 1642–1643. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Young, D.C.; Garrett, P.M.; Owen, D.A.; Schwerin, S.G.; Tebbe, F.N.; Wegner, P.A. Preparation and characterization of the (3)-1,2- and (3)-1,7-dicarbadodecahydroundecaborate(-1) ions. J. Am. Chem. Soc. 1968, 90, 862–868. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Young, D.C.; Andrews, T.D.; Howe, D.V.; Pilling, R.L.; Pitts, A.D.; Reintjer, M.; Warren, L.F.; Wegner, P.A. π-Dicarbollyl derivatives of the transition metals. Metallocene analogs. J. Am. Chem. Soc. 1968, 90, 879–896. [Google Scholar] [CrossRef]
- Grimes, R.N. Transition metal metallacarbaboranes. In Comprehensive Organometallic Chemistry II.; Elsevier: Oxford, UK, 1995; Volume 1, pp. 373–430. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Chemistry of cobalt bis(dicarbollides). A review. Collect. Czech. Chem. Commun. 1999, 64, 783–805. [Google Scholar] [CrossRef]
- Grimes, R.N. Metallacarboranes in the new millennium. Coord. Chem. Rev. 2000, 200, 773–811. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Chemistry of nickel and iron bis(dicarbollides). A review. J. Organomet. Chem. 2002, 614–615, 27–36. [Google Scholar] [CrossRef]
- Hosmane, N.S.; Maguire, J.A. Metallacarboranes of d- and f-block metals. In Comprehensive Organometallic Chemistry III; Elsevier: Oxford, UK, 2007; Volume 3, pp. 175–264. [Google Scholar] [CrossRef]
- Grimes, R.N. Metallacarboranes of the transition and lanthanide elements. In Carboranes, 3rd ed.; Academic Press: London, UK, 2016; pp. 711–903. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Swain, B.R.; Mahanta, C.S.; Jena, B.B.; Hosmane, N.S. Cobalt bis(dicarbollide) anion and its derivatives. J. Organomet. Chem. 2017, 849–850, 170–194. [Google Scholar] [CrossRef]
- Kar, S.; Pradhan, A.N.; Ghosh, S. Polyhedral metallaboranes and metallacarboranes. In Comprehensive Organometallic Chemistry IV; Elsevier: Oxford, UK, 2022; Volume 9, pp. 263–369. [Google Scholar] [CrossRef]
- Pak, R.H.; Primus, F.J.; Rickard-Dickson, K.J.; Ng, L.L.; Kane, R.R.; Hawthorne, M.F. Preparation and properties of nido-carborane-specific monoclonal antibodies for potential use in boron neutron capture therapy for cancer. Proc. Natl. Acad. Sci. USA 1995, 92, 6986–6990. [Google Scholar] [CrossRef] [Green Version]
- Hogenkamp, H.P.C.; Collins, D.A.; Live, D.; Benson, L.M.; Naylor, S. Synthesis and characterization of nido-carborane-cobalamin conjugates. Nucl. Med. Biol. 2000, 27, 89–92. [Google Scholar] [CrossRef]
- Nakamura, H.; Miyajima, Y.; Takei, T.; Kasaoka, S.; Maruyama, K. Synthesis and vesicle formation of a nido-carborane cluster lipid for boron neutron capture therapy. Chem. Commun. 2004, 17, 1910–1911. [Google Scholar] [CrossRef]
- Yinghuai, Z.; Peng, A.T.; Carpenter, K.; Maguire, J.A.; Hosmane, N.S.; Takagaki, M. Substituted carborane-appended water-soluble single-wall carbon nanotubes: New approach to boron neutron capture therapy drug delivery. J. Am. Chem. Soc. 2005, 127, 9875–9880. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, Y.; Nakamura, H.; Kuwata, Y.; Lee, J.-D.; Masunaga, S.; Ono, K.; Maruyama, K. Transferrin-loaded nido-carborane liposomes: Tumor-targeting boron delivery system for neutron capture therapy. Bioconjug. Chem. 2006, 17, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Takagaki, M.; Bode, B.P.; Snajdr, I.; Patel, D.; Sharman, C.; Bux, M.; Bux, S.; Kotora, M.; Hosmane, N.S. Carborane-appended saccharides: Prime candidates for boron neutron capture therapy (BNCT) clinical trials. Biochem. Biophys. J. Neutron Ther. Cancer Treat. 2013, 1, 15–21. [Google Scholar]
- Pietrangeli, D.; Rosa, A.; Pepe, A.; Altieri, S.; Bortolussi, S.; Postuma, I.; Protti, N.; Ferrari, C.; Cansolino, L.; Clerici, A.M.; et al. Water-soluble carboranyl-phthalocyanines for BNCT. Synthesis, characterization, and in vitro tests of the Zn(II)-nido-carboranyl-hexylthiophthalocyanine. Dalton Trans. 2015, 44, 11021–11028. [Google Scholar] [CrossRef]
- Lee, W.; Sarkar, S.; Ahn, H.; Kim, J.Y.; Lee, Y.J.; Chang, Y.; Yoo, J. PEGylated liposome encapsulating nido-carborane showed significant tumor suppression in boron neutron capture therapy (BNCT). Biochem. Biophys. Res. Commun. 2020, 522, 669–675. [Google Scholar] [CrossRef]
- Tolmachev, V.; Bruskin, A.; Sjöberg, S.; Carlsson, J.; Lundqvist, H. Preparation, radioiodination and in vitro evaluation of a nido-carborane-dextran conjugate, a potential residualizing label for tumor targeting proteins and peptides. J. Radioanal. Nucl. Chem. 2004, 261, 107–112. [Google Scholar] [CrossRef]
- Winberg, K.J.; Persson, M.; Malmström, P.-U.; Sjöberg, S.; Tolmachev, V. Radiobromination of anti-HER2/neu/ErbB-2 monoclonal antibody using the p-isothiocyanatobenzene derivative of the [76Br]undecahydro-bromo-7,8-dicarba-nido-undecaborate(1-) ion. Nucl. Med. Biol. 2004, 31, 425–433. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Chyan, M.-K.; Hamlin, D.K.; Kegley, B.B.; Risler, R.; Pathare, P.M.; Quinn, J.; Vessella, R.L.; Foulon, C.; Zalutsky, M.; et al. Reagents for astatination of biomolecules: Comparison of the in vivo distribution and stability of some radioiodinated/astatinated benzamidyl and nido-carboranyl compounds. Bioconjug. Chem. 2004, 15, 203–223. [Google Scholar] [CrossRef]
- El-Zaria, M.E.; Genady, A.R.; Janzen, N.; Petlura, C.I.; Beckford Vera, D.R.; Valliant, J.F. Preparation and evaluation of carborane-derived inhibitors of prostate specific membrane antigen (PSMA). Dalton Trans. 2014, 43, 4950–4961. [Google Scholar] [CrossRef]
- Wilkinson, S.M.; Gunosewoyo, H.; Barron, M.L.; Boucher, A.; McDonnell, M.; Turner, P.; Morrison, D.E.; Bennett, M.R.; McGregor, I.S.; Rendina, L.M.; et al. The first CNS-active carborane: A novel P2X7 receptor antagonist with antidepressant activity. ACS Chem. Neurosci. 2014, 5, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Neumann, W.; Xu, S.; Sárosi, M.B.; Scholz, M.S.; Crews, B.C.; Ghebreselasie, K.; Banerjee, S.; Marnett, L.J.; Hey-Hawkins, E. nido-Dicarbaborate induces potent and selective inhibition of cyclooxygenase-2. ChemMedChem 2016, 11, 175–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Różycka, D.; Korycka-Machała, M.; Żaczek, A.; Dziadek, J.; Gurda, D.; Orlicka-Płocka, M.; Wyszko, E.; Biniek-Antosiak, K.; Rypniewski, W.; Olejniczak, A.B. Novel isoniazid-carborane hybrids active in vitro against Mycobacterium tuberculosis. Pharmaceuticals 2020, 13, 465. [Google Scholar] [CrossRef] [PubMed]
- Useini, L.; Mojić, M.; Laube, M.; Lönnecke, P.; Dahme, J.; Sárosi, M.B.; Mijatović, S.; Maksimović-Ivanić, D.; Pietzsch, J.; Hey-Hawkins, E. Carboranyl analogues of mefenamic acid and their biological evaluation. ACS Omega 2022, 7, 24282–24291. [Google Scholar] [CrossRef]
- Nghia, N.V.; Oh, J.; Jung, J.; Lee, M.H. Deboronation-induced turn-on phosphorescent sensing of fluorides by iridium(III) cyclometalates with o-carborane. Organometallics 2017, 36, 2573–2580. [Google Scholar] [CrossRef]
- Nghia, N.V.; Oh, J.; Sujith, S.; Jung, J.; Lee, M.H. Tuning the photophysical properties of carboranyl luminophores by closo- to nido-carborane conversion and application to OFF–ON fluoride sensing. Dalton Trans. 2018, 47, 17441–17449. [Google Scholar] [CrossRef]
- Sujith, S.; Nam, E.B.; Lee, J.; Lee, S.U.; Lee, M.H. Enhancing the thermally activated delayed fluorescence of nido-carborane-appended triarylboranes by steric modification of the phenylene linker. Inorg. Chem. Front. 2020, 7, 3456–3464. [Google Scholar] [CrossRef]
- Kim, M.; Im, S.; Ryu, C.H.; Lee, S.H.; Hong, J.H.; Lee, K.M. Impact of deboronation on the electronic characteristics of closo-o-carborane: Intriguing photophysical changes in triazole-appended carboranyl luminophores. Dalton Trans. 2021, 50, 3207–3215. [Google Scholar] [CrossRef]
- Lee, S.H.; Mun, M.S.; Kim, M.; Lee, J.H.; Hwang, H.; Lee, W.; Lee, K.M. Alteration of intramolecular electronic transition via deboronation of carbazole-based o-carboranyl compound and intriguing ‘turn-on’ emissive variation. RSC Adv. 2021, 11, 24057–24064. [Google Scholar] [CrossRef]
- Alconchel, A.; Crespo, O.; García-Orduña, P.; Gimeno, M.C. closo- or nido-Carborane diphosphane as responsible for strong thermochromism or time activated delayed fluorescence (TADF) in [Cu(N^N)(P^P)]0/+. Inorg. Chem. 2021, 60, 18521–18528. [Google Scholar] [CrossRef]
- Uemura, K.; Tanaka, K.; Chujo, Y. Conformation-dependent electron donation of nido-carborane substituents and its influence on phosphorescence of tris(2,2′-bipyridyl)ruthenium(II) complex. Crystals 2022, 12, 688. [Google Scholar] [CrossRef]
- Teixidor, F.; Nuñez, R.; Viñas, C.; Sillanpää, R.; Kivekäs, R. Contribution of the nido-[7,8-C2B9H10]- anion to the chemical stability, basicity, and 31P NMR chemical shift in nido-o-carboranylmonophosphines. Inorg. Chem. 2001, 40, 2587–2594. [Google Scholar] [CrossRef]
- Timofeev, S.V.; Zakharova, M.V.; Mosolova, E.M.; Godovikov, I.A.; Ananyev, I.V.; Sivaev, I.B.; Bregadze, V.I. Tungsten carbonyl σ-complexes of nido-carborane thioethers. J. Organomet. Chem. 2012, 721–722, 92–96. [Google Scholar] [CrossRef]
- Kazakov, G.S.; Sivaev, I.B.; Suponitsky, K.Y.; Kirilin, A.D.; Bregadze, V.I.; Welch, A.J. Facile synthesis of closo-nido bis(carborane) and its highly regioselective halogenation. J. Organomet. Chem. 2016, 805, 1–5. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Erokhina, S.A.; Sivaev, I.B.; Bregadze, V.I. Synthesis of C-methoxy- and C,C’-dimethoxy-ortho-carboranes. J. Organomet. Chem. 2020, 927, 121523. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Maguire, J.A.; Hosmane, N.S. Polyhedral boron clusters in materials science. New J. Chem. 2011, 35, 1955–1972. [Google Scholar] [CrossRef]
- Green, J.; Mayer, N. Thermal stability of carborane-containing polymers. J. Macromol. Sci. A Chem. 1967, 1, 135–145. [Google Scholar] [CrossRef]
- Zhang, X.; Kong, L.; Dai, L.; Zhang, X.; Wang, Q.; Tan, Y.; Zhang, Z. Synthesis, characterization, and thermal properties of poly(siloxane-carborane)s. Polymer 2011, 52, 4777–4784. [Google Scholar] [CrossRef]
- Kolel-Veetil, M.K.; Dominguez, D.D.; Klug, C.A.; Fears, K.P.; Qadri, S.B.; Fragiadakis, D.; Keller, T.M. Hybrid inorganic–organic Poly(carborane-siloxane-arylacetylene) structural isomers with in-chain aromatics: Synthesis and properties. J. Polym. Sci. A Polym. Chem. 2013, 51, 2638–2650. [Google Scholar] [CrossRef]
- Nuñez, R.; Romero, I.; Teixidor, F.; Viñas, C. Icosahedral boron clusters: A perfect tool for the enhancement of polymer features. Chem. Soc. Rev. 2016, 45, 5147–5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Feng, C.; Yang, J.; Chen, G. High thermally stable thermosetting polyimides derived from a carborane-containing tetramine. High Perform. Polym. 2019, 31, 548–556. [Google Scholar] [CrossRef]
- Liu, F.; Fang, G.; Yang, H.; Yang, S.; Zhang, X.; Zhang, Z. Carborane-containing aromatic polyimide films with ultrahigh thermo-oxidative stability. Polymers 2019, 11, 1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Gao, M.; Zhao, L.; Zhao, Y.; Li, T.; Chen, K.; Hu, X.; He, L.; Huang, Q.; Liu, M.; et al. Recent advances in carborane-siloxane polymers. React. Func. Polym. 2022, 173, 105213. [Google Scholar] [CrossRef]
- Minyaylo, E.O.; Kudryavtseva, A.I.; Zubova, V.Y.; Anisimov, A.A.; Zaitsev, A.V.; Ol’shevskaya, V.A.; Dolgushin, F.M.; Peregudov, A.S.; Muzafarov, A.M. Synthesis of mono- and polyfunctional organosilicon derivatives of polyhedral carboranes for the preparation of hybrid polymer materials. New J. Chem. 2022, 46, 11143–11148. [Google Scholar] [CrossRef]
- Tsuboya, N.; Lamrani, M.; Hamasaki, R.; Ito, M.; Mitsuishi, M.; Miyashita, T.; Yamamoto, Y. Nonlinear optical properties of novel carborane–ferrocene conjugated dyads. Electron-withdrawing characteristics of carboranes. J. Mater. Chem. 2002, 12, 2701–2705. [Google Scholar] [CrossRef]
- Yan, J.-F.; Zhu, G.-G.; Yuan, Y.; Lin, C.-X.; Huang, S.-P.; Yuan, Y.-F. Carborane bridged ferrocenyl conjugated molecules: Synthesis, structure, electrochemistry and photophysical properties. New J. Chem. 2020, 44, 7569–7576. [Google Scholar] [CrossRef]
- Lee, S.; Shin, J.; Ko, D.-H.; Han, W.-S. A new type of carborane-based electron-accepting material. Chem. Commun. 2020, 84, 12741–12744. [Google Scholar] [CrossRef]
- Ochi, J.; Tanaka, K.; Chujo, Y. Recent progress in the development of solid-state luminescent o-carboranes with stimuli responsivity. Angew. Chem. Int. Ed. 2020, 59, 9841–9855. [Google Scholar] [CrossRef]
- Yi, S.; Kim, M.; Ryu, C.H.; You, D.K.; Seo, Y.J.; Lee, K.M. Relationship between the molecular geometry and the radiative efficiency in naphthyl-based bis-ortho-carboranyl luminophores. Molecules 2022, 27, 6565. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Wegner, P.A. Reconstruction of the 1,2-dicarbaclovododecaborane(12) structure by boron-atom insertion with (3)-1,2-dicarbollide ions. J. Am. Chem. Soc. 1968, 90, 896–901. [Google Scholar] [CrossRef]
- Yamazaki, H.; Ohta, K.; Endo, Y. Regioselective synthesis of triiodo-o-carboranes and tetraiodo-o-carborane. Tetrahedron Lett. 2005, 46, 3119–3122. [Google Scholar] [CrossRef]
- Teixidor, F.; Barberà, G.; Viñas, C.; Sillanpää, R.; Kivekäs, R. Synthesis of boron-iodinated o-carborane derivatives. Water stability of the periodinated monoprotic salt. Inorg. Chem. 2006, 45, 3496–3498. [Google Scholar] [CrossRef] [PubMed]
- Barbera, G.; Viñas, C.; Teixidor, F.; Welch, A.J.; Rosair, G.M. Retention of the B(3)-X (X = Br, I) bond in closo-o-carborane derivatives after nucleophilic attack. The first synthesis of [3-X-7-R-7,8-nido-C2B9H10]- (X = Br, I). Crystal structure of [HNMe3][3-I-7,8-nido-C2B9H11]. J. Organomet. Chem. 2002, 657, 217–223. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Li, T.C.; Farha, O.K.; Machan, C.W.; She, C.; Stern, C.L.; Marks, T.J.; Hupp, J.T.; Mirkin, C.A. Electronic tuning of nickel-based bis(dicarbollide) redox shuttles in dye-sensitized solar cells. Angew. Chem. Int. Ed. 2010, 49, 5339–5343. [Google Scholar] [CrossRef]
- Safronov, A.V.; Shlyakhtina, N.I.; Hawthorne, M.F. New approach to the synthesis of 3-alkyl-1,2-dicarba-closo-dodecaboranes: Reaction of alkyldichloroboranes with thallium dicarbollide. Organometallics 2012, 31, 2764–2769. [Google Scholar] [CrossRef]
- Safronov, A.V.; Shlyakhtina, N.I.; Everett, T.A.; VanGordon, M.R.; Sevryugina, Y.V.; Jalisatgi, S.S.; Hawthorne, M.F. Direct observation of bis(dicarbollyl)nickel conformers in solution by fluorescence spectroscopy: An approach to redox-controlled metallacarborane molecular motors. Inorg. Chem. 2014, 53, 10045–10053. [Google Scholar] [CrossRef]
- Shlyakhtina, N.I.; Safronov, A.V.; Sevryugina, Y.V.; Jalisatgi, S.S.; Hawthorne, M.F. Synthesis, characterization, and preliminary fluorescence study of a mixed-ligand bis(dicarbollyl)nickel complex bearing a tryptophan-BODIPY FRET couple. J. Organomet. Chem. 2015, 798, 234–244. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Ol’shevskaya, V.A.; Sulaimankulova, D.D.; Antonovich, V.A. Cleavage of 3-amino-o-carborane and its’ N-derivatives by bases into the 3-amino-7,8-dicarbaundecaborate anion and its N-derivatives. Russ. Chem. Bull. 1991, 40, 1026–1032. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Shmal’ko, A.V.; Stogniy, M.Y.; Suponitsky, K.Y.; Sivaev, I.B. Isomeric ammonio derivatives of nido-carborane 3- and 10-H3N-7,8-C2B9H11. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 901–904. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Telegina, A.A.; Levit, G.L.; Krasnov, V.P. N-Aminoacyl-3-amino-nido-carboranes as a group of boron-containing derivatives of natural amino acids. J. Org. Chem. 2022, 87, 5437–5441. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Ol’shevskaya, V.A.; Sulaimankulova, D.D. Synthesis of 3-isocyano-nido-7,8-dicarbaundecaborate salts and their use as new isonitrile ligands in transition-metal complexes. Russ. Chem. Bull. 1993, 42, 1395–1397. [Google Scholar] [CrossRef]
- Shmalko, A.V.; Anufriev, S.A.; Stogniy, M.Y.; Suponitsky, K.Y.; Sivaev, I.B. Synthesis and structure of 3-arylazo derivatives of ortho-carborane. New J. Chem. 2020, 44, 10199–10202. [Google Scholar] [CrossRef]
- Lebedev, V.N.; Balagurova, E.V.; Zakharkin, L.I. Destruction of B-polyfluorosubstituted o-carboranes into anions of B-fluorosubstituted nido-7,8-dicarbaundecaborates by the action of ethanolic alkali and amines. Russ. Chem. Bull. 1995, 44, 1102–1106. [Google Scholar] [CrossRef]
- Brattsev, V.A.; Knyazev, S.P.; Danilova, G.N.; Vostrikova, T.N.; Stanko, V.I. Intramolecular nucleophilic cleavage of 3-oxy-1,2- and 2-oxy-1,7-dicarbaclosododecaboranes(12). Russ. J. Gen. Chem. 1976, 46, 2627. [Google Scholar]
- Zakharkin, L.I.; Kalinin, V.N.; Gedymin, V.V. Synthesis and some reactions of 3-amino-o-carboranes. J. Organomet. Chem. 1969, 16, 371–379. [Google Scholar] [CrossRef]
- Kasar, R.A.; Knudsen, G.M.; Kahl, S.B. Synthesis of 3-amino-1-carboxy-o-carborane and an improved, general method for the synthesis of all three C-amino-C-carboxycarboranes. Inorg. Chem. 1999, 38, 2936–2940. [Google Scholar] [CrossRef] [Green Version]
- Valliant, J.F.; Schaffer, P. A new approach for the synthesis of isonitrile carborane derivatives.: Ligands for metal based boron neutron capture therapy (BNCT) and boron neutron capture synovectomy (BNCS) agents. J. Inorg. Biochem. 2001, 85, 43–51. [Google Scholar] [CrossRef]
- Zhao, D.; Xie, Z. [3-N2-o-C2B10H11][BF4]: A useful synthon for multiple cage boron functionalizations of o-carborane. Chem. Sci. 2016, 7, 5635–5639. [Google Scholar] [CrossRef] [Green Version]
- Au, Y.K.; Zhang, J.; Quan, Y.; Xie, Z. Ir-Catalyzed selective B(3)-H amination of o-carboranes with NH3. J. Am. Chem. Soc. 2021, 143, 4148–4153. [Google Scholar] [CrossRef]
- Cheng, R.; Qiu, Z.; Xie, Z. Iridium-catalysed regioselective borylation of carboranes via direct B–H activation. Nat. Commun. 2017, 8, 14827. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Liao, X.; Hartwig, J.F. Meta halogenation of 1,3-disubstituted arenes via iridium-catalyzed arene borylation. J. Am. Chem. Soc. 2007, 129, 15434–15435. [Google Scholar] [CrossRef]
- Zhang, G.; Lv, G.; Li, L.; Chen, F.; Cheng, J. Copper-catalyzed halogenation of arylboronic acids. Tetrahedron Lett. 2011, 52, 1993–1995. [Google Scholar] [CrossRef]
- Ren, Y.-L.; Tian, X.-Z.; Dong, C.; Zhao, S.; Wang, J.; Yan, M.; Qi, X.; Liu, G. A simple and effective copper catalyst for the conversion of arylboronic acids to aryl iodides at room temperature. Catal. Commun. 2013, 32, 15–17. [Google Scholar] [CrossRef]
- Molloy, J.J.; O’Rourke, K.M.; Frias, C.P.; Sloan, N.L.; West, M.J.; Pimlott, S.L.; Sutherland, A.; Watson, A.J.B. Mechanism of Cu-catalyzed aryl boronic acid halodeboronation using electrophilic halogen: Development of a base-catalyzed iododeboronation for radiolabeling applications. Org. Lett. 2019, 21, 2488–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Hynes, J. Copper-catalyzed chlorination of functionalized arylboronic acids. Org. Lett. 2020, 12, 1192–1195. [Google Scholar] [CrossRef]
- Bardakov, V.G.; Yakubenko, A.A.; Verkhov, V.A.; Antonov, A.S. Organoboron derivatives of 1,8-bis(dimethylamino)naphthalene: Synthesis, structure, stability, and reactivity. Organometallics 2022, 41, 1501–1508. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Timofeev, S.V.; Zhidkova, O.B.; Suponitsky, K.Y.; Sivaev, I.B. Synthesis, crystal structure, and some transformations of 9,12-dichloro-ortho-carborane. Crystals 2022, 12, 1251. [Google Scholar] [CrossRef]
- Barbera, G.; Vaca, A.; Teixidor, F.; Sillanpää, R.; Kivekäs, R.; Viñas, C. Designed synthesis of new ortho-carborane derivatives: From mono- to polysubstituted frameworks. Inorg. Chem. 2008, 47, 7309–7316. [Google Scholar] [CrossRef]
- Zefirov, Y.V.; Zorky, P.M. New applications of van der Waals radii in chemistry. Russ. Chem. Rev. 1995, 64, 415–428. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals; Butterworth Heinemann: Burlington, MA, USA, 2009. [Google Scholar]
- APEX2 and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3,6-Cl2-1,2-C2B10H10 (3) | Cs [3-Cl-7,8-C2B9H11] (11) | |
|---|---|---|
| Formula | C2H10B10Cl2 | Cs+C2B9H11Cl− |
| FW | 213.10 | 300.76 |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | C2/c | Pbca |
| a, Å | 14.746(7) | 10.693(2) |
| b, Å | 6.805(4) | 11.149(2) |
| c, Å | 11.485(6) | 18.174(4) |
| β, deg | 115.259(14) | 90 |
| V, Å3 | 1042.4(9) | 2166.6(8) |
| Z | 4 | 8 |
| ρcalc, g·cm−3 | 1.358 | 1.844 |
| F(000) | 424 | 1120 |
| μ, mm−1 | 0.557 | 3.599 |
| θ range, deg | 3.06–26.08 | 2.24–26.15 |
| Independent reflections | 1030 | 2141 |
| Completeness to theta θ, % | 99.0 | 98.8 |
| Refined parameters | 85 | 122 |
| GOF (F2) | 0.984 | 1.037 |
| Reflections with I > 2σ(I) | 587 | 1549 |
| R1(F) (I > 2σ(I)) a | 0.0592 | 0.0579 |
| wR2(F2) (all data) b | 0.1473 | 0.1382 |
| Largest diff. peak/hole, e·Å−3 | 0.400/−0.478 | 0.977/−1.299 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shmal’ko, A.V.; Anufriev, S.A.; Suponitsky, K.Y.; Sivaev, I.B. How to Protect ortho-Carborane from Decapitation—Practical Synthesis of 3,6-Dihalogen Derivatives 3,6-X2-1,2-C2B10H10 (X = Cl, Br, I). Inorganics 2022, 10, 207. https://doi.org/10.3390/inorganics10110207
Shmal’ko AV, Anufriev SA, Suponitsky KY, Sivaev IB. How to Protect ortho-Carborane from Decapitation—Practical Synthesis of 3,6-Dihalogen Derivatives 3,6-X2-1,2-C2B10H10 (X = Cl, Br, I). Inorganics. 2022; 10(11):207. https://doi.org/10.3390/inorganics10110207
Chicago/Turabian StyleShmal’ko, Akim V., Sergey A. Anufriev, Kyrill Yu. Suponitsky, and Igor B. Sivaev. 2022. "How to Protect ortho-Carborane from Decapitation—Practical Synthesis of 3,6-Dihalogen Derivatives 3,6-X2-1,2-C2B10H10 (X = Cl, Br, I)" Inorganics 10, no. 11: 207. https://doi.org/10.3390/inorganics10110207
APA StyleShmal’ko, A. V., Anufriev, S. A., Suponitsky, K. Y., & Sivaev, I. B. (2022). How to Protect ortho-Carborane from Decapitation—Practical Synthesis of 3,6-Dihalogen Derivatives 3,6-X2-1,2-C2B10H10 (X = Cl, Br, I). Inorganics, 10(11), 207. https://doi.org/10.3390/inorganics10110207