

Combining a Low Valent Molybdenum(0) Center with a Strongly σ-Donating Mesoionic Carbene Chelate Ligand—Synthesis and Structural Characterization


Abstract
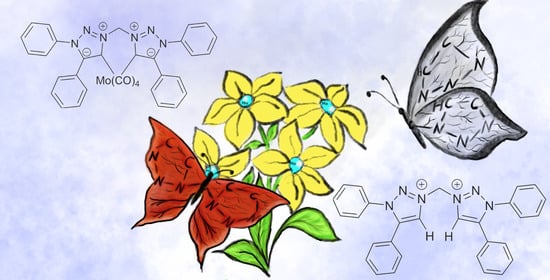
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of the Target Compounds
2.2. X-ray Crystallography
2.3. Spectroscopic Results
2.4. Electrochemical Investigations
3. Materials and Methods
3.1. General Considerations
3.2. SCXRD
3.3. DFT Calculations
3.4. Synthetic Procedures
3.4.1. 1,5-Diphenyl-1H-1,2,3-triazole (1)
3.4.2. Synthesis of 3,3′-Methylenebis(1,5-diphenyl-1H-1,2,3-triazol-3-ium) triflate (2)
3.4.3. Synthesis of [Mo(CO)4(bta)] (3)
- (A)
- Preparation of [Mo(CO)4(bta)] (3) by transmetalation with silver oxide
- (B)
- Preparation of [Mo(CO)4(bta)] (3) in a one-pot synthesis using [Mo(CO)4(piperidine)2] under reflux conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Baskin, J.M.; Prescher, J.A.; Lo, A.; Bertozzi, C.R. A comparative study of bioorthogonal reactions with azides. ACS Chem. Biol. 2006, 1, 644–648. [Google Scholar] [CrossRef]
- Huisgen, R.; Knorr, R.; Möbius, L.; Szeimies, G. 1.3-Dipolare Cycloadditionen, XXIII. Einige Beobachtungen zur Addition organischer Azide an CC-Dreifachbindungen. Chem. Ber. 2006, 98, 4014–4021. [Google Scholar] [CrossRef]
- Kwok, S.W.; Fotsing, J.R.; Fraser, R.J.; Rodionov, V.O.; Fokin, V.V. Transition-metal-free catalytic synthesis of 1,5-diaryl-1,2,3-triazoles. Org. Lett. 2010, 12, 4217–4219. [Google Scholar] [CrossRef] [Green Version]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V.V. Ruthenium-catalyzed azide-alkyne cycloaddition: Scope and mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar] [CrossRef]
- Huisgen, R.; Szeimies, G.; Möbius, L. 1.3-Dipolare Cycloadditionen, XXXII. Kinetik der Additionen organischer Azide an CC-Mehrfachbindungen. Chem. Ber. 2006, 100, 2494–2507. [Google Scholar] [CrossRef]
- Schulze, B.; Schubert, U.S. Beyond click chemistry—supramolecular interactions of 1,2,3-triazoles. Chem. Soc. Rev. 2014, 43, 2522. [Google Scholar] [CrossRef] [PubMed]
- Mathew, P.; Neels, A.; Albrecht, M. 1,2,3-Triazolylidenes as versatile abnormal carbene ligands for late transition metals. J. Am. Chem. Soc. 2008, 130, 13534–13535. [Google Scholar] [CrossRef] [Green Version]
- Maity, R.; Sarkar, B. Chemistry of Compounds Based on 1,2,3-Triazolylidene-Type Mesoionic Carbenes. JACS Au 2022, 2, 22–57. [Google Scholar] [CrossRef]
- Suntrup, L.; Hohloch, S.; Sarkar, B. Expanding the Scope of Chelating Triazolylidenes: Mesoionic Carbenes from the 1,5-Click-Regioisomer and Catalytic Synthesis of Secondary Amines from Nitroarenes. Chem. Eur. J. 2016, 22, 18009–18018. [Google Scholar] [CrossRef] [PubMed]
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Hettmanczyk, L.; Manck, S.; Hoyer, C.; Hohloch, S.; Sarkar, B. Heterobimetallic complexes with redox-active mesoionic carbenes as metalloligands: Electrochemical properties, electronic structures and catalysis. Chem. Commun. 2015, 51, 10949–10952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettmanczyk, L.; Suntrup, L.; Klenk, S.; Hoyer, C.; Sarkar, B. Heteromultimetallic Complexes with Redox-Active Mesoionic Carbenes: Control of Donor Properties and Redox-Induced Catalysis. Chem. Eur. J. 2017, 23, 576–585. [Google Scholar] [CrossRef]
- Vanicek, S.; Podewitz, M.; Stubbe, J.; Schulze, D.; Kopacka, H.; Wurst, K.; Muller, T.; Lippmann, P.; Haslinger, S.; Schottenberger, H.; et al. Highly Electrophilic, Catalytically Active and Redox-Responsive Cobaltoceniumyl and Ferrocenyl Triazolylidene Coinage Metal Complexes. Chemistry 2018, 24, 3742–3753. [Google Scholar] [CrossRef]
- Woods, J.A.; Lalrempuia, R.; Petronilho, A.; McDaniel, N.D.; Müller-Bunz, H.; Albrecht, M.; Bernhard, S. Carbene iridium complexes for efficient water oxidation: Scope and mechanistic insights. Energy Environ. Sci. 2014, 7, 2316–2328. [Google Scholar] [CrossRef] [Green Version]
- van der Meer, M.; Glais, E.; Siewert, I.; Sarkar, B. Electrocatalytic Dihydrogen Production with a Robust Mesoionic Pyridylcarbene Cobalt Catalyst. Angew. Chem. Int. Ed. 2015, 54, 13792–13795. [Google Scholar] [CrossRef]
- Suntrup, L.; Stein, F.; Klein, J.; Wilting, A.; Parlane, F.G.L.; Brown, C.M.; Fiedler, J.; Berlinguette, C.P.; Siewert, I.; Sarkar, B. Rhenium Complexes of Pyridyl-Mesoionic Carbenes: Photochemical Properties and Electrocatalytic CO2 Reduction. Inorg. Chem. 2020, 59, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Kleinhans, G.; Guisado-Barrios, G.; Liles, D.C.; Bertrand, G.; Bezuidenhout, D.I. A rhodium(I)-oxygen adduct as a selective catalyst for one-pot sequential alkyne dimerization-hydrothiolation tandem reactions. Chem. Commun. 2016, 52, 3504–3507. [Google Scholar] [CrossRef] [Green Version]
- Baschieri, A.; Monti, F.; Matteucci, E.; Mazzanti, A.; Barbieri, A.; Armaroli, N.; Sambri, L. A Mesoionic Carbene as Neutral Ligand for Phosphorescent Cationic Ir(III) Complexes. Inorg. Chem. 2016, 55, 7912–7919. [Google Scholar] [CrossRef]
- Chabera, P.; Liu, Y.; Prakash, O.; Thyrhaug, E.; Nahhas, A.E.; Honarfar, A.; Essen, S.; Fredin, L.A.; Harlang, T.C.; Kjaer, K.S.; et al. A low-spin Fe(III) complex with 100-ps ligand-to-metal charge transfer photoluminescence. Nature 2017, 543, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Hettmanczyk, L.; Spall, S.J.P.; Klenk, S.; van der Meer, M.; Hohloch, S.; Weinstein, J.A.; Sarkar, B. Structural, Electrochemical, and Photochemical Properties of Mono- and Digold(I) Complexes Containing Mesoionic Carbenes. Eur. J. Inorg. Chem. 2017, 2017, 2112–2121. [Google Scholar] [CrossRef] [Green Version]
- Matteucci, E.; Monti, F.; Mazzoni, R.; Baschieri, A.; Bizzarri, C.; Sambri, L. Click-Derived Triazolylidenes as Chelating Ligands: Achievement of a Neutral and Luminescent Iridium(III)-Triazolide Complex. Inorg. Chem. 2018, 57, 11673–11686. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, B.; Suntrup, L. Illuminating Iron: Mesoionic Carbenes as Privileged Ligands in Photochemistry. Angew. Chem. Int. Ed. 2017, 56, 8938–8940. [Google Scholar] [CrossRef] [PubMed]
- Vivancos, A.; Segarra, C.; Albrecht, M. Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications. Chem. Rev. 2018, 118, 9493–9586. [Google Scholar] [CrossRef] [PubMed]
- Guisado-Barrios, G.; Soleilhavoup, M.; Bertrand, G. 1H-1,2,3-Triazol-5-ylidenes: Readily Available Mesoionic Carbenes. Acc. Chem. Res. 2018, 51, 3236–3244. [Google Scholar] [CrossRef]
- Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Bis(1,2,3-triazol-5-ylidenes) (i-bitz) as Stable 1,4-Bidentate Ligands Based on Mesoionic Carbenes (MICs). Organometallics 2011, 30, 6017–6021. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.F.; Olivares, M.; Vivancos, Á.; Guisado-Barrios, G.; Albrecht, M.; Royo, B. (Di)triazolylidene manganese complexes in catalytic oxidation of alcohols to ketones and aldehydes. Catal. Sci. Technol. 2019, 9, 2421–2425. [Google Scholar] [CrossRef]
- Friaes, S.; Realista, S.; Gomes, C.S.B.; Martinho, P.N.; Veiros, L.F.; Albrecht, M.; Royo, B. Manganese complexes with chelating and bridging di-triazolylidene ligands: Synthesis and reactivity. Dalton Trans. 2021, 50, 5911–5920. [Google Scholar] [CrossRef]
- Suntrup, L.; Klenk, S.; Klein, J.; Sobottka, S.; Sarkar, B. Gauging Donor/Acceptor Properties and Redox Stability of Chelating Click-Derived Triazoles and Triazolylidenes: A Case Study with Rhenium(I) Complexes. Inorg. Chem. 2017, 56, 5771–5783. [Google Scholar] [CrossRef]
- Vivancos, Á.; Albrecht, M. Influence of the Linker Length and Coordination Mode of (Di)Triazolylidene Ligands on the Structure and Catalytic Transfer Hydrogenation Activity of Iridium(III) Centers. Organometallics 2017, 36, 1580–1590. [Google Scholar] [CrossRef]
- Hohloch, S.; Suntrup, L.; Sarkar, B. Arene–Ruthenium(II) and −Iridium(III) Complexes with “Click”-Based Pyridyl-triazoles, Bis-triazoles, and Chelating Abnormal Carbenes: Applications in Catalytic Transfer Hydrogenation of Nitrobenzene. Organometallics 2013, 32, 7376–7385. [Google Scholar] [CrossRef]
- Sluijter, S.N.; Elsevier, C.J. Synthesis and Reactivity of Heteroditopic Dicarbene Rhodium(I) and Iridium(I) Complexes Bearing Chelating 1,2,3-Triazolylidene–Imidazolylidene Ligands. Organometallics 2014, 33, 6389–6397. [Google Scholar] [CrossRef]
- Bolje, A.; Kosmrlj, J. A selective approach to pyridine appended 1,2,3-triazolium salts. Org. Lett. 2013, 15, 5084–5087. [Google Scholar] [CrossRef] [PubMed]
- Bernet, L.; Lalrempuia, R.; Ghattas, W.; Mueller-Bunz, H.; Vigara, L.; Llobet, A.; Albrecht, M. Tunable single-site ruthenium catalysts for efficient water oxidation. Chem. Commun. 2011, 47, 8058–8060. [Google Scholar] [CrossRef]
- Delgado-Rebollo, M.; Canseco-Gonzalez, D.; Hollering, M.; Mueller-Bunz, H.; Albrecht, M. Synthesis and catalytic alcohol oxidation and ketone transfer hydrogenation activity of donor-functionalized mesoionic triazolylidene ruthenium(II) complexes. Dalton Trans. 2014, 43, 4462–4473. [Google Scholar] [CrossRef] [Green Version]
- Suntrup, L.; Stein, F.; Hermann, G.; Kleoff, M.; Kuss-Petermann, M.; Klein, J.; Wenger, O.S.; Tremblay, J.C.; Sarkar, B. Influence of Mesoionic Carbenes on Electro- and Photoactive Ru and Os Complexes: A Combined (Spectro-)Electrochemical, Photochemical, and Computational Study. Inorg. Chem. 2018, 57, 13973–13984. [Google Scholar] [CrossRef]
- Leigh, V.; Ghattas, W.; Lalrempuia, R.; Muller-Bunz, H.; Pryce, M.T.; Albrecht, M. Synthesis, photo-, and electrochemistry of ruthenium bis(bipyridine) complexes comprising a N-heterocyclic carbene ligand. Inorg. Chem. 2013, 52, 5395–5402. [Google Scholar] [CrossRef]
- Stubbe, J.; Neuman, N.I.; McLellan, R.; Sommer, M.G.; Nossler, M.; Beerhues, J.; Mulvey, R.E.; Sarkar, B. Isomerization Reactions in Anionic Mesoionic Carbene-Borates and Control of Properties and Reactivities in the Resulting Co(II) Complexes through Agostic Interactions. Angew. Chem. Int. Ed. 2021, 60, 499–506. [Google Scholar] [CrossRef]
- Suntrup, L.; Beerhues, J.; Etzold, O.; Sarkar, B. Copper(I) complexes bearing mesoionic carbene ligands: Influencing the activity in catalytic halo-click reactions. Dalton Trans. 2020, 49, 15504–15510. [Google Scholar] [CrossRef]
- Hidai, M.; Tominari, K.; Uchida, Y. Preparation and properties of dinitrogen-molybdenum complexes. J. Am. Chem. Soc. 1972, 94, 110–114. [Google Scholar] [CrossRef]
- Ohki, Y.; Aoyagi, K.; Seino, H. Synthesis and Protonation of N-Heterocyclic-Carbene-Supported Dinitrogen Complexes of Molybdenum(0). Organometallics 2015, 34, 3414–3420. [Google Scholar] [CrossRef]
- Beerhues, J.; Aberhan, H.; Streit, T.-N.; Sarkar, B. Probing Electronic Properties of Triazolylidenes through Mesoionic Selones, Triazolium Salts, and Ir-Carbonyl-Triazolylidene Complexes. Organometallics 2020, 39, 4557–4564. [Google Scholar] [CrossRef]
- Elvers, B.J.; Sawall, M.; Oberem, E.; Heckenberger, K.; Ludwig, R.; Neymeyr, K.; Schulzke, C.; Krewald, V.; Fischer, C. Towards operando IR- and UV-vis-Spectro-Electrochemistry: A Comprehensive Matrix Factorisation Study on Sensitive and Transient Molybdenum and Tungsten Mono-Dithiolene Complexes. Chemistry-Methods 2021, 1, 22–35. [Google Scholar] [CrossRef]
- Beerhues, J.; Fauche, K.; Cisnetti, F.; Sarkar, B.; Gautier, A. A dicopper(I)-dimesoionic carbene complex as a click catalyst: Mechanistic implications. Dalton Trans. 2019, 48, 8931–8936. [Google Scholar] [CrossRef]
- Hsu, M.-A.; Yeh, W.-Y.; Chiang, M.Y. Syntheses, characterization and structures of CpFe(CO)(μ-I)(μ-dppm)M(CO)4 (M=Cr, Mo, W). J. Organomet. Chem. 1998, 552, 135–143. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Kump, R.L. A convenient synthesis of cis-Mo(CO)4L2 derivatives (L = Group 5a ligand) and a qualitative study of their thermal reactivity toward ligand dissociation. Inorg. Chem. 1978, 17, 2680–2682. [Google Scholar] [CrossRef]
- Wieland, S.; van Eldik, R. Mechanistic Study of the Substitution Behavior of Complexes of the Type M(CO),(THF) (M = Cr, Mo, W). Organometallics 1991, 10, 3110–3114. [Google Scholar] [CrossRef]
- Elvers, B.J.; Schulzke, C.; Fischer, C. Photochemical Unmasking of 1,3-Dithiol-2-ones: An Alternative Route to Heteroleptic Dithiolene Complexes from Low-Valent Molybdenum and Tungsten Precursors. Eur. J. Inorg. Chem. 2019, 2019, 2796–2805. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Poulain, A.; Canseco-Gonzalez, D.; Hynes-Roche, R.; Müller-Bunz, H.; Schuster, O.; Stoeckli-Evans, H.; Neels, A.; Albrecht, M. Synthesis and Tunability of Abnormal 1,2,3-Triazolylidene Palladium and Rhodium Complexes. Organometallics 2011, 30, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Navarro, M.; Wang, S.; Müller-Bunz, H.; Redmond, G.; Farràs, P.; Albrecht, M. Triazolylidene Metal Complexes Tagged with a Bodipy Chromophore: Synthesis and Monitoring of Ligand Exchange Reactions. Organometallics 2017, 36, 1469–1478. [Google Scholar] [CrossRef]
- Sureshbabu, B.; Ramkumar, V.; Sankararaman, S. Facile base-free in situ generation and palladation of mesoionic and normal N-heterocyclic carbenes at ambient conditions. Dalton Trans. 2014, 43, 10710–10712. [Google Scholar] [CrossRef]
- Hettmanczyk, L.; Schmid, B.; Hohloch, S.; Sarkar, B. Palladium(ii)-Acetylacetonato Complexes with Mesoionic Carbenes: Synthesis, Structures and Their Application in the Suzuki-Miyaura Cross Coupling Reaction. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic carbenes: Synthesis and coordination chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, T.; Tao, X.; Huang, Y. 2,2,6,6-Tetramethylpiperidinium triflate (TMPT): A highly selective and self-separated catalyst for esterification. Tetrahedron Lett. 2016, 57, 4905–4909. [Google Scholar] [CrossRef]
- Suntrup, L.; Kleoff, M.; Sarkar, B. Serendipitous discoveries of new coordination modes of the 1,5-regioisomer of 1,2,3-triazoles enroute to the attempted synthesis of a carbon-anchored tri-mesoionic carbene. Dalton Trans. 2018, 47, 7992–8002. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Stein, F.; Kirsch, M.; Beerhues, J.; Albold, U.; Sarkar, B. Mono- and Di-Mesoionic Carbene-Boranes: Synthesis, Structures and Utility as Reducing Agents. Eur. J. Inorg. Chem. 2021, 2021, 2417–2424. [Google Scholar] [CrossRef]
- Bouffard, J.; Keitz, B.K.; Tonner, R.; Lavallo, V.; Guisado-Barrios, G.; Frenking, G.; Grubbs, R.H.; Bertrand, G. Synthesis of Highly Stable 1,3-Diaryl-1H-1,2,3-triazol-5-ylidenes and their Applications in Ruthenium-Catalyzed Olefin Metathesis. Organometallics 2011, 30, 2617–2627. [Google Scholar] [CrossRef]
- Arduengo, A.J. Looking for Stable Carbenes: The Difficulty in Starting Anew. Acc. Chem. Res. 1999, 32, 913–921. [Google Scholar] [CrossRef]
- Burling, S.; Field, L.D.; Li, H.L.; Messerle, B.A.; Turner, P. Mononuclear Rhodium(I) Complexes with Chelating N-Heterocyclic Carbene Ligands − Catalytic Activity for Intramolecular Hydroamination. Eur. J. Inorg. Chem. 2003, 2003, 3179–3184. [Google Scholar] [CrossRef]
- Dierkes, P.; van Leeuwen, P.W.N.M. The bite angle makes the difference: A practical ligand parameter for diphosphine ligands. J. Chem. Soc., Dalton Trans. 1530. [Google Scholar] [CrossRef]
- Gradert, C.; Krahmer, J.; Sonnichsen, F.D.; Nather, C.; Tuczek, F. Small-Molecule Activation with Molybdenum(0) Complexes Supported by Mixed Imidazol-2-Ylidene/Phosphanyl Hybrid Ligands—Electronic and Structural Consequences of Substituting a Phosphane by a Carbene Group. Eur. J. Inorg. Chem. 2013, 2013, 3943–3955. [Google Scholar] [CrossRef]
- Gradert, C.; Krahmer, J.; Sönnichsen, F.D.; Näther, C.; Tuczek, F. Molybdenum(0)–carbonyl complexes supported by mixed benzimidazol-2-ylidene/phosphine ligands: Influence of benzannulation on the donor properties of the NHC groups. J. Organomet. Chem. 2014, 770, 61–68. [Google Scholar] [CrossRef]
- Lappert, M.F.; Pye, P.L.; McLaughlin, G.M. Carbene complexes. Part 9. Electron-rich olefin-derived carbene–molybdenum(0) and amidinium molybdate(0) complexes, and the crystal and molecular structure of cis-tetracarbonylbis(1,3-dimethylimidazolidin-2-ylidene)molybdenum(0), cis-[Mo(CO)4{CN(Me)CH2CH2NMe}2]. J. Chem. Soc., Dalton Trans. 1977, 1272–1282. [Google Scholar] [CrossRef]
- Bens, T.; Boden, P.; Di Martino-Fumo, P.; Beerhues, J.; Albold, U.; Sobottka, S.; Neuman, N.I.; Gerhards, M.; Sarkar, B. Chromium(0) and Molydenum(0) Complexes with a Pyridyl-Mesoionic Carbene Ligand: Structural, (Spectro)electrochemical, Photochemical, and Theoretical Investigations. Inorg. Chem. 2020, 59, 15504–15513. [Google Scholar] [CrossRef]
- Maitra, K.; Nelson, J.H. Formation of 1,2-bis (diphenylphosphino)ethane, dppe, from two coordinated diphenylvinylphosphine (DPVP) ligands crystal structures of cis- and trans-(DPVP)2Mo(CO)4 and (dppe)M(CO)4 (M=Cr, Mo). Polyhedron 1998, 18, 203–210. [Google Scholar] [CrossRef]
- Braga, S.S.; Coelho, A.C.; Gonçalves, I.S.; Almeida Paz, F.A. (2,2′-Bipyridine)tetracarbonylmolybdenum(0). Acta Crystallogr. E: Crystallogr. Commun. 2007, 63, m780–m782. [Google Scholar] [CrossRef]
- Öfele, K.; Herrmann, W.A.; Mihalios, D.; Elison, M.; Herdtweck, E.; Priermeier, T.; Kiprof, P. Heterocyclische Carbene IV. Metallkomplexe mit heterocyclischen Carben-Liganden: Synthese, Struktur, Strukturdynamik. J. Organomet. Chem. 1995, 498, 1–14. [Google Scholar] [CrossRef]
- Clark, M.L.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Electrocatalytic CO2 reduction by M(bpy-R)(CO)4 (M = Mo, W; R = H, tBu) complexes. Electrochemical, spectroscopic, and computational studies and comparison with group 7 catalysts. Chem. Sci. 2014, 5, 1894–1900. [Google Scholar] [CrossRef] [Green Version]
- Macholdt, H.-T.; Elias, H. Ligand Substitution in Molybdenum(0) Carbonyl Complexes Mo(CO)5(amine) and cis -Mo(CO)4(amine)2: Kinetics and High-Pressure Effects. Inorg. Chem. 1984, 23, 4315–4321. [Google Scholar] [CrossRef]
- Bock, H.; tom Dieck, H. Metall(0)-Verbindungen mit nichtaromatischen Stickstoff-π-Systemen, VI. 1.4-Diaza-butadien-molybdän-tetracarbonyle: Synthesen, Eigenschaften und Bindungsmodell. Chem. Ber. 1967, 100, 228–246. [Google Scholar] [CrossRef]
- Öfele, K.; Roos, E.; Herberhold, M. Isomerisierungsreaktionen von cis- und trans-Dicarben-Komplexen des Typs M(CO)4L2 (M = Cr, Mo, W). Z. Naturforsch. B 1976, 31, 1070–1077. [Google Scholar] [CrossRef]
- Heinze, J. Cyclic Voltammetry—“Electrochemical Spectroscopy”. Angew. Chem. Int. Ed. 1984, 23, 831–847. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Label | (1) | (2) | Bond Label | (3) |
|---|---|---|---|---|
| N1-N2 | 1.3554(18) | 1.322(2) | N1-N2 | 1.320(4) |
| N2-N3 | 1.3127(19) | 1.318(2) | N2-N3 | 1.336(4) |
| N1-C1 | 1.362(2) | 1.373(3) | N1-C5 | 1.369(5) |
| N3-C2 | 1.356(2) | 1.347(3) | N3-C6 | 1.366(5) |
| N3-C3 | - | 1.463(2) | N3-C7 | 1.448(4) |
| C2-H2 | 0.98(3) | 0.93(2) | C6-Mo | 2.276(4) |
| C1-C2 | 1.375(2) | 1.364(3) | C5-C6 | 1.392(5) |
| N5-N6 | - | 1.322(2) | N5-N6 | 1.321(4) |
| N4-C4 | - | 1.347(3) | N4-C8 | 1.366(5) |
| C4-H4 | - | 0.90(2) | C8-Mo | 2.284(3) |
| N1-N2-N3 | 106.80(13) | 103.70(15) | N1-N2-N3 | 101.5(3) |
| N4-N5-N6 | - | 103.29(16) | N6-N5-N4 | 101.6(2) |
| N3-C2-C1 | 109.27(14) | 105.88(18) | N3-C6-C5 | 100.5(3) |
| N4-C4-C5 | - | 105.71(18) | N4-C8-C9 | 100.2(3) |
| N3-C2-H2 | 123.0(15) | 123.6(13) | N3-C6-Mo | 120.8(2) |
| N4-C4-H4 | - | 121.2(14) | N4-C8-Mo | 120.5(2) |
| C1-C2-H2 | 127.8(15) | 130.5(13) | C5-C6-Mo | 137.2(3) |
| C5-C4-H4 | - | 133.1(15) | C9-C8-Mo | 138.7(3) |
| N3-C3-N4 | - | 108.98(16) | N3-C7-N4 | 109.0(3) |
| C2-C3-C4 | - | 104.31(2) | C6-C7-C8 | 74.751(13) |
| L^L= | bta | Iet^Iet | bIet^bIet | (Ime)2 a | PyMIC | dppe | bipy |
|---|---|---|---|---|---|---|---|
| Ref. | This Work | [64] | [65] | [66] | [67] | [68] | [69] |
| Mo-L1 | 2.276(4) | 2.261(4) | 2.249(2) | 2.293(3) | 2.202(1) | 2.500(2) | 2.249(3) |
| Mo-L2 | 2.284(3) | 2.259(4) | 2.257(2) | 2.293(3) | 2.275(1) | 2.495(2) | 2.240(3) |
| Mo-COax. | 2.030(4) | 2.036(6) | 2.041(3) | 2.032(3) | 2.051(2) | 2.053(9) | 2.056(4) |
| 2.018(3) | 2.024(6) | 2.031(3) | 2.024(3) | 2.040(2) | 2.030(9) | 2.023(4) | |
| Mo-COeq. | 1.986(3) | 1.988(5) | 2.000(3) | 1.981(3) | 1.997(2) | 1.999(8) | 1.962(4) |
| 1.961(4) | 1.977(6) | 1.986(3) | 1.979(3) | 1.973(1) | 1.974(8) | 1.952(4) | |
| C-Oax. | 1.146(5) | 1.143(6) | 1.143(3) | 1.146(4) | 1.149(2) | 1.143(9) | 1.150(4) |
| 1.142(4) | 1.134(6) | 1.137(4) | 1.144(4) | 1.146(29 | 1.120(9) | 1.134(4) | |
| C-Oeq. | 1.156(5) | 1.157(6) | 1.150(3) | 1.157(3) | 1.158(2) | 1.164(9) | 1.168(4) |
| 1.152(4) | 1.149(6) | 1.145(4) | 1.156(3) | 1.156(2) | 1.140(8) | 1.160(4) | |
| L1-Mo-L2 | 83.60(14) | 79.6(2) | 80.37(9) | 85.64(10) | 72.83(4) | 80.2(1) | 72.36(10) |
| L1-Mo-CO | 175.54(15) | 173.3(2) | 175.49(11) | 178.98(10) | 168.22(5) | 171.4(2) | 171.37(12) |
| L2-Mo-CO | 177.4(2) | 175.1(2) | 176.01(10) | 178.50(10) | 169.71(5) | 170.6(6) | 170.66(12) |
| COax.-Mo-COax. | 172.77(19) | 170.2(2) | 170.42(10) | 169.82(11) | 171.77(6) | 176.6(3) | 167.83(13) |
| L^L= | bta | I^I | Iet^Iet | bIet^bIet | (Ime)2 | T^T | PyMIC | dppe | bipy | phen | pDAB |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | This Work a | [70] b | [64] c | [65] c | [66] b | [70] b | [67] d | [49] b | [71] e | [72] a | [73] f |
| ν1(CO) | 1990 | 1994 | 1987 | 1994 | 1994 | 2005 | 2006 | 2018 | 2016 | 2010 | 2016 |
| ν2(CO) | 1864 | 1872 | 1876 | 1875 | 1868 | 1884 | 1896 | 1923 | 1904 | 1915 | 1901 |
| ν3(CO) | 1810 | 1841 | 1818 | 1860 | 1863 | 1853 | 1876 | 1896 | 1877 | 1875 | 1869 |
| ν4(CO) | 1799 | 1817 | 1838 | 1830 | 1884 | 1832 | 1835 | 1832 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elvers, B.J.; Schulan, P.; Pätsch, S.; Fischer, C.; Schulzke, C. Combining a Low Valent Molybdenum(0) Center with a Strongly σ-Donating Mesoionic Carbene Chelate Ligand—Synthesis and Structural Characterization. Inorganics 2022, 10, 216. https://doi.org/10.3390/inorganics10110216
Elvers BJ, Schulan P, Pätsch S, Fischer C, Schulzke C. Combining a Low Valent Molybdenum(0) Center with a Strongly σ-Donating Mesoionic Carbene Chelate Ligand—Synthesis and Structural Characterization. Inorganics. 2022; 10(11):216. https://doi.org/10.3390/inorganics10110216
Chicago/Turabian StyleElvers, Benedict Josua, Paul Schulan, Sebastian Pätsch, Christian Fischer, and Carola Schulzke. 2022. "Combining a Low Valent Molybdenum(0) Center with a Strongly σ-Donating Mesoionic Carbene Chelate Ligand—Synthesis and Structural Characterization" Inorganics 10, no. 11: 216. https://doi.org/10.3390/inorganics10110216
APA StyleElvers, B. J., Schulan, P., Pätsch, S., Fischer, C., & Schulzke, C. (2022). Combining a Low Valent Molybdenum(0) Center with a Strongly σ-Donating Mesoionic Carbene Chelate Ligand—Synthesis and Structural Characterization. Inorganics, 10(11), 216. https://doi.org/10.3390/inorganics10110216