1. Introduction
The oxidation of KA oil (a mixture of cyclohexanol and cyclohexanone) in the presence of small amounts of metal complexes is reported to yield adipic acid (AA) [
1,
2]. The oxidation reaction usually takes place in chemical industries at high temperatures, and requires a two-step oxidation of cyclohexane or a one-step oxidation of cyclohexanol. The chemical industries also make use of nitric acid, which contributes to global warming, acid rain, and depletion of the ozone layer [
3,
4,
5]. Moreover, advanced oxidation processes, which include oxidizing cyclohexane under UV light [
6], oxidizing cyclohexane under an oxygen gas medium [
7], and carbonizing catalytic materials derived from lignocellulosic biomass also known as lignin-derived bio-oils [
8], provide efficient and energy-conserving ways to prepare AA. According to reports, the bulk of these oxidation reactions are notably unsatisfactory, as low yields in AA are being achieved. The alternative use of aqueous hydrogen peroxide (H
2O
2) was related to a sustainable process, and has been widely adopted. Hydrogen peroxide slowly decomposes in a catalytic system, to form water as a byproduct at low temperatures [
9,
10].
Alcañiz-Monge, et al. [
11], in 2014 reported the use of homogeneous (HPW, HPMo) and different heterogenous catalysts (PCoMo, PNiMo, PFeMo VPMo, (N(C
3H
7)
4)
3PMo
12O
40, (NH
4)
3PMo
12O
40, Cs
3PW
12O
40, and (NH4)
3PMoO
4)) for AA preparation with cyclohexene and 30% hydrogen peroxide; the reaction was performed in a batch reactor bed at 75 °C under autogenous pressure. The most active catalyst was found to be Cs
3PW
12O
40, as it gave an 80% yield of AA with acetic acid, while a 13% yield of AA was obtained in the absence of acetic acid. Moreover, Jianhui, et al. [
12], developed a catalytic system with β-Anderson manganohexamolybdate containing a single-atom Mn active site; to enhance the AA preparation in a 30% H
2O
2 medium, cyclohexanone was adopted as the starting material. Considerable high conversion, selectivity, and turnover numbers (TON) (99%, 97%, and 2427.5 hr
−1) of AA were obtained.
The utilization of coordination compounds, such as metal complexes, and coordination polymers (metal–organic frameworks) for highly effective catalytic reactions, has been reported [
13,
14,
15,
16,
17,
18,
19,
20,
21]. Coordination compounds are various types of compounds with metal ions—such as alkali and alkaline earth metals, transitional metals, and lanthanide(III) ions—coordinated by various functional groups, such as carboxylate, hydroxyl, and amine groups [
21]. Several coordination compounds have shown intriguing catalytic activities owing to their flexibility, accessibility to surface acidity, possession of strong bonds between di- and trivalent metal ions and ligands pillared by nitrogen and oxygen atoms, superior stability, and resistance to decomposition upon heating. Reports, however, have revealed that the oxidation of cyclohexanone yields AA in pure form using coordination compounds [
1,
9,
16].
Furthermore, the global demand on adipic usage in chemical industries for the production of nylon-6,6, plasticizers, etc., is increasing [
1,
10]. The need to obtain AA in high yield and pure form is therefore imperative. Based on this premise, two new metal complexes, [Mn(2,6-pydc)
2](imi) (
1) and [Co(H
2pza)
2(H
2O)
2(NO
3)]•NO
3 (
2), were prepared as catalysts for the oxidation of cyclohexanone to AA in high yield and pure form. Both of the compounds utilized were chosen from Mn(II) ion, Co(II) ion, pyridylcarboxylate, azolate, and pyrazinamide ligands. The choice of ligands employed influenced the length, porosity, rigidity, and coordination modes on the resulting frameworks of coordination polymer material. These ligands consisted of adjustable linkers suitable for generating lower-dimensional coordination polymers. The synthesized materials were categorized into heterogeneous compounds, and as they exhibited good thermal behaviors, we selected them as being suitable for oxidizing a substrate like adipic acid [
21]. Solvents were chosen from aqueous H
2O
2 and acetonitrile, because the yields of the products were between 60% and 90% in other literatures [
20,
21]. The effects of reaction temperature, time, and oxidants on the catalytic reactions, and the mechanism, kinetics, and thermodynamics of the reaction were studied in this research work.
2. Materials and Methods
All the reagents and solvents used in this study were of analytical grade, and required no further purification. Mn(II) ion salt, Co(II) ion salt, pyridylcarboxylate, imidazole, and pyrazinamide ligands were purchased from Sigma–Aldrich (USA) and Tokyo Chemical Industry (China). All the chemicals were used as received. FTIR and UV-visible spectra analyses were recorded on a SHIMADZU (Tokyo) FTIR-8501 spectrophotometer and a SHIMADZU UV-1650pc UV-VIS spectrophotometer. XRD analyses were performed on a PANanalytical X’pert Pro dual goniometer diffractometer at 294 K, using CuKα radiation (λ = 1.54059). Thermogravimetric analysis (TGA) of the complexes was performed on a Seiko (USA) DTA/TGA 320 instrument under air atmosphere at a heating rate of 10 °C min−1 from 30 to 600 °C. The % metal content of both catalysts was determined by EDAX (energy-dispersive absorption X-ray spectroscopy) analysis on an EDAX AMETEK (USA) Z2 Analyzer. HPLC analyses of the oxidation products and side products were carried out using a 1260 Infinity HPLC instrument made by Agilent Technologies (Santa Clara, CA, USA).
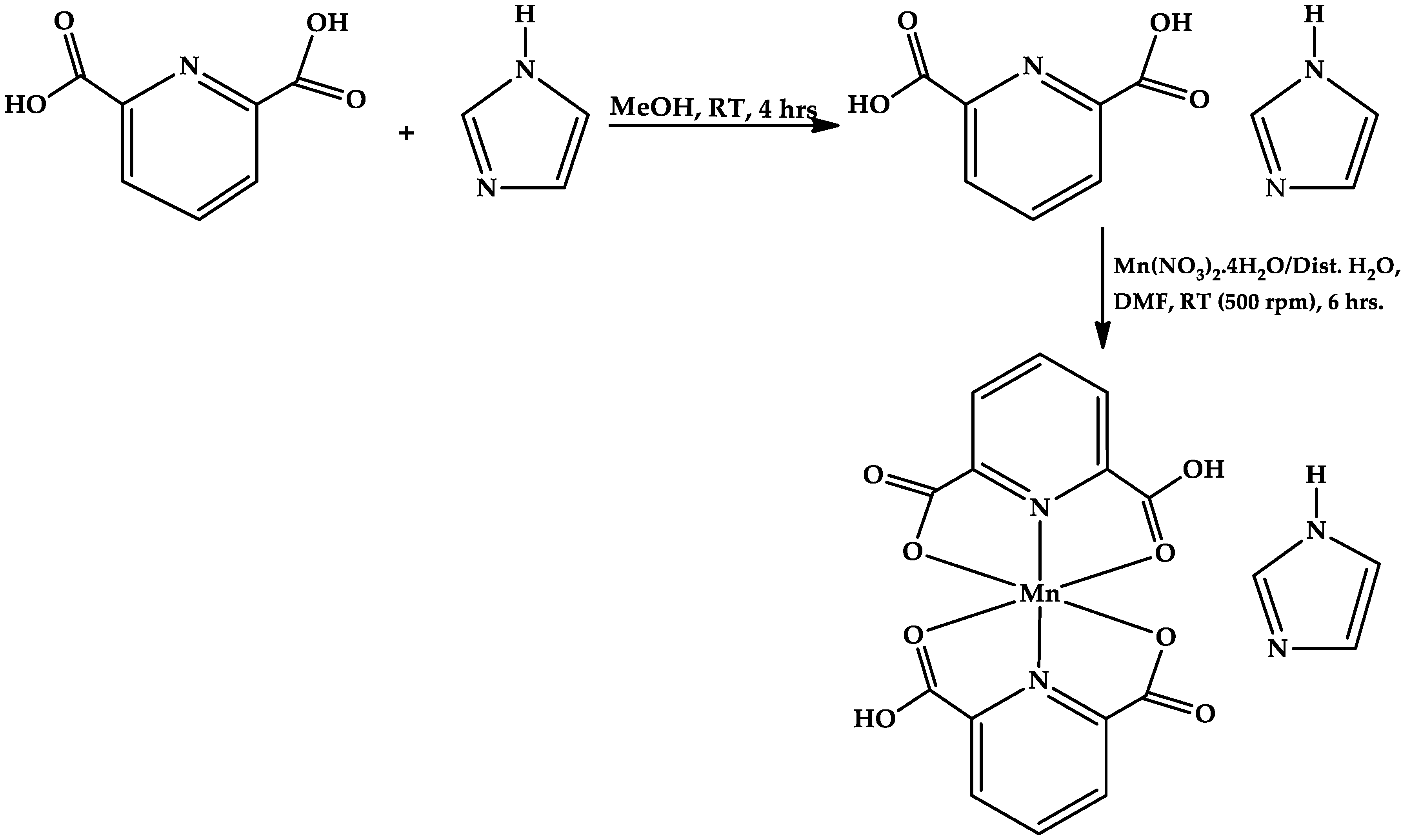
Synthesis of the co-crystals of (2,6-pydc–imi
+): The synthetic procedure described by Gross, et al. [
5], was modified and adopted for the synthesis of the co-crystals of (2,6-pydc–imi
+). In a typical reaction, 2,6-pyridinedicarboxylic acid (2,6-pydc) (167.12 mg, 1 mmol) and imidazole (imi) (68.078 mg, 1 mmol) were dissolved separately in 25 mL beakers with 5 mL methanolic solution; thereafter, the two solutions were gently mixed together in a 25 mL beaker for about 1 min. The final solution was transferred to a 100 mL round-bottom flask, and stirred (at 500 rpm) for 4 h at room temperature (RT). The clear filtrate obtained was allowed to settle in a clean 20 mL vial, and then kept in the dark to allow formation of crystals. After 7 days, plate-like colorless co-crystals, suitable for single-crystal X-ray diffraction analysis, were formed (
Scheme 1). The co-crystals were filtered off, washed with methanol, and dried in air for approximately 10 min.
Synthesis of [Mn(2,6-pydc)
2](imi): The synthetic procedure described by Gross, et al. [
5], was modified and adopted for the synthesis of [Mn(2,6-pydc)
2](imi). In practice, co-crystals of (2,6-pydc–imi
+) (250 mg) were mechanochemically crushed to powder form, and later poured into a 20 mL vial where 5 mL DMF solution was added; the resulting solution was kept for 3 days in the dark; thereafter, manganese dinitrate tetrahydrate, Mn(NO
3)
2.4H
2O (251.01 mg, 1 mmol), already pre-dissolved with distilled water (5 mL) was added, and manually stirred for about 10 min. The mixture was transferred to a 100 mL round-bottom flask, and continuously stirred (500 rpm) for 6 h at room temperature (RT); the obtained grey filtrate was kept in the dark without any disturbance, for crystal growth to occur. Flake-like grey crystals suitable for single-crystal X-ray diffraction analysis were formed after 16 days. (
Scheme 1). The mother liquor was decanted, and the crystals were filtered off, washed with ethanol, and dried in air for about 10 min. Yield: 60.2%, Mp. = 501 °C, Anal. Calc. for C
17H
10MnN
4O
8 (%): C, 43.43; H, 2.08; N, 7.24. Found %: C, 43.12; H, 2.41; N, 7.32. IR (KBr, ν/cm
−1): 1319, 1692, 1394, 1456, 1312, 663, 578. UV-Vis. (solid state diffuse-reflectance) (λ
LMCT, nm): 317; (λ
d-d, nm): 687.
Synthesis of [Co(H
2pza)
2(H
2O)
2(NO
3)]•NO
3: The synthetic procedure described by Raja, et al. [
14], was modified and adopted for the synthesis of [Co(H
2pza)
2(H
2O)
2(NO
3)].NO
3. In a typical reaction, pyrazinamide (H
2pza) (246.23 mg, 2 mmol) and cobalt dinitrate hexahydrate, Co(NO
3)
2.6H
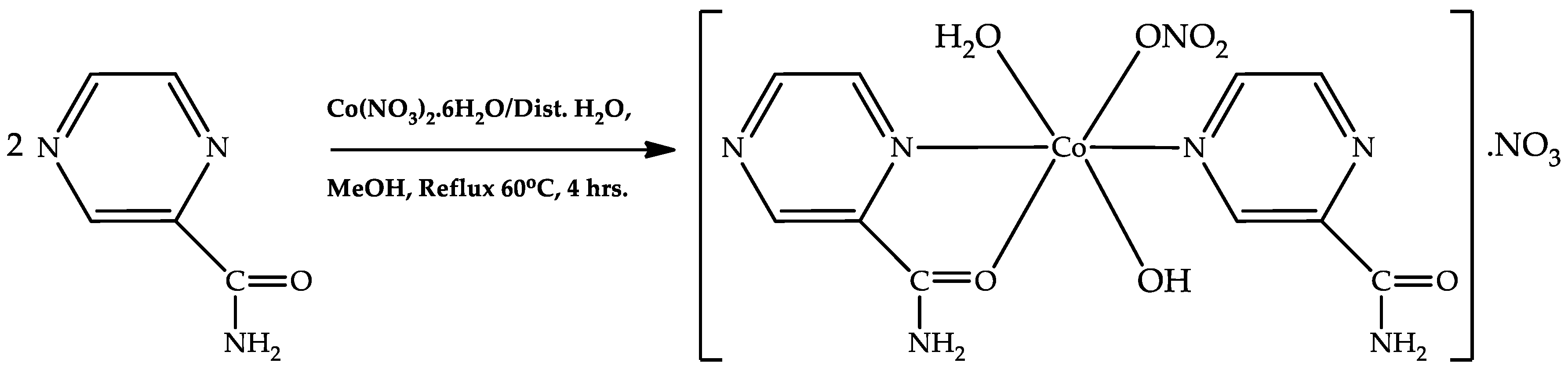
2O (290.70 mg, 1 mmol), were dissolved separately in a 25 mL beaker with 5 mL methanolic solution and 5 mL distilled water. The two solutions were gently mixed together for about 30 s, and the mixture was refluxed in a 100 mL round-bottom flask at 60 °C for 4 h. Filtrate obtained from the refluxing mixture was allowed to stand freely on the bench without any disturbance for crystal growth formation. Block-like pink crystals, suitable for single-crystal X-ray diffraction analysis, were formed after 4 days (
Scheme 2). The mother liquor was decanted. and the pink crystals were filtered off, washed with methanol, and dried in air (10 min). Yield: 52.5%, Mp. = 479 °C, Anal. Calc. for C
10H
14CoN
8O
10 (%): C, 28.66; H, 3.50; N, 24.34. Found %: C, 31.02; H, 3.58; N, 22.65. IR (KBr, ν/cm
−1): 1190, 1684, 782, 675. UV-Vis. (solid state diffuse reflectance) (λ
LLCT/LMCT, nm): 236, 272, 333; (λ
d-d, nm): 525.
3. Results and Discussion
The flake-like grey crystals of [Mn(2,6-pydc)
2](imi) (
1) were synthesized from the co-crystals of (2,6-pydc–imi
+) via mechanochemical crushing, followed by adopting the room temperature synthetic method to obtain the product (
Scheme 1); the yield was 60.2%. The block-like pink crystals of [Co(H
2pza)
2(H
2O)
2(NO
3)].NO
3 (
2) were synthesized in good yield (52.2%) by refluxing pyrazinamide (H
2pza) and cobalt dinitrate hexahydrate with suitable solvent (
Scheme 2); the stability temperatures, 287 °C and 140 °C, and melting point values, 501 °C and >470 °C, indicated their stability at high temperatures (see ESI for TGA estimation). The % metal content values (75.58% and 24.78%) was evaluated using MP-AES (microwave plasma-absorption emission spectroscopy), and predicted the presence of active metals (Mn and Co), respectively.
The physicochemical properties of
1 and
2 are summarized in
Table 1.
The FTIR spectrum result for 2,6-pydc showed a peak at 1181 cm
−1 characteristic of the ν(C-N) band, and had shifted to 1319 cm
−1 in the spectrum of compound
1 (see
Figure S1). This suggested the coordination of imine nitrogen to Mn(II) ion. The low-intensity peaks at 1709 cm
−1, 1256 cm
−1, and 3212 cm
−1 were characteristic of ν(C=O), ν(C-O), and ν(OH) bands in 2,6-pydc, and had shifted to 1692 cm
−1 and 1394 cm
−1 in the spectrum of compound
1, confirming the coordination of the carboxylate oxygen atoms to the Mn(II) ion [
21]. The hydrogen bond interactions between neighboring bonded atoms, i.e., imi and 2,6-pydc bearing N-H and OH groups, were confirmed by the disappearance of an ν(OH) stretching band and the appearance of an ν(C-N) weak band at 1295 cm
−1, respectively; these bands differed from those in the spectrum of
1 [
22,
23]. The separation (∆νCOO, 164) between ν
sCOO and ν
asCOO suggested that the carboxylate groups of 2,6-pydc exhibited bidentate bonding mode [
21]. The characteristic peaks for the coordinated metal ions (νMn-O and νMn-N bands) appeared at 663 cm
−1 and 578 cm
−1 (
Table S1).
For complex
2, the FTIR spectrum of H
2pza displayed peaks at 1253 cm
−1 and 1708 cm
−1 characteristic of ν(C-N) and ν(C=O) stretching bands, and had shifted to 1190 cm
−1 in the spectrum of
2 (see
Figure S2); this indicated the involvement of Co(II) ion in coordination with pyrimidine nitrogen and carbonyl oxygen of H
2pza [
24]. Furthermore, the two broad peaks, 3285 cm
−1 and 3301 cm
−1—characteristic of trace amounts of water, ν(OH
w), and amine ν(NH
2)
sy/asy stretching bands in the spectrum of the free ligand—had completely disappeared in the spectrum of
2, suggesting the presence of hydrogen bonds between the NH
2 group, the coordinated H
2O, and the uncoordinated nitrate in
2 [
24,
25,
26]. The peaks for the coordinated Co
2+ ion, 782 cm
−1 and 675 cm
−1, were characteristic of ν(Co-O) and ν(Co-N) bonds (
Table S2).
The electronic spectrum for imi and 2,6-pydc showed three absorption bands, 230 nm, 277 nm and 280 nm, which were characteristic of imine nitrogen (C=N), carbonyl oxygen (C=O), and hydroxyl (OH) groups’ chromophores within the cyclopentadiene and benzene rings of the parent ligands. These bands were assigned to π → π* and n → π* transitions. A shift to higher wavelengths, 317 nm and 687 nm, was observed near the visible region in the spectrum of compound
1 (
Figure S3), probably due to ligand-to-metal charge transfer (LMCT) [
22,
23]. The band at 687 nm was attributed to a weak spin-forbidden transition,
4T
1g ←
6A
1g (see
Table S3), due to complexation between the Mn
2+ ion and the O^N donor atoms of 2,6-pydc [
21].
For complex
2, the electronic spectrum of the ligand (H
2pza) which exhibited absorption bands between 221–316 nm had shifted to 236–333 nm in the spectrum of
2 (
Figure S4). These bands were characteristic of the imine (C=N), amine (NH
2), and carbonyl (C=O) groups’ chromophores attached to the H
2pza benzene ring, and had been assigned to π → π* and n → π* transitions. The significant shifts in the spectrum of
2 near the visible region were due to ligand-to-metal charge transfer (LMCT). We tentatively assigned two low-intensity bands at 236 nm and 272 nm in the spectrum of
2 to a mixed LLCT/LMCT, due to less contributions of O^N donor groups in H
2pza in the excited state [
25]. In addition, the hyperchromic shift, 525 nm, observed at the visible region in the spectrum of the complex, was assigned to
4A
2g →
4T
1g (P) transition (see
Table S4), due to d-d* transition of the d
7 system of Co(II) cation. The effect of the hyperchromic shift was attributed to complexation between Co
2+ and H
2pza.
Thermogravimetric analysis was conducted under nitrogen atmosphere, at a maximum temperature of 600 °C. The TG curve of
1 (
Figure S5) indicated a starting stepwise weight loss in the range of 228–357 °C, which corresponded to the decomposition of uncoordinated imi moiety (mass loss was 43.1%/44.8% for calc./found). A second-stage weight loss was in the range of 366–505 °C, which corresponded to the decomposition of 2,6-pydc bound to Mn(II) ion in the chemical structure (mass loss was 17.5%/20.3% for calc./found). The final stage of thermal decomposition was between 506–600 °C, which was attributed to MnO residue formation. The thermal decomposition behavior of 2 (
Figure S6) also revealed a weight loss in the range of 125 °C to 365 °C, which corresponded to: (i) the decomposition of uncoordinated (lattice) nitrate molecules; (ii) coordinated nitrate molecules; and (iii) coordinated water molecules. The mass loss of nitrate(s) was found to be 5.3/10.3% (calc./found), while that of water molecule(s) was given as 4.4/5.6% (calc./found). The mass loss which occurred in the range of 366 °C and 471 °C was attributed to the decomposition of the H
2pza organic specie; experimental mass loss was found to be 34.0/30.5% (found/calc.). The final stage of thermal decomposition started from 472 °C down to 600 °C, and was ascribed to the formation of CoO residue (metal oxide). The thermal stability (>228 °C and >128 °C) observed in
1 and
2 suggested that both compounds would serve as good catalyst materials, and these results were comparable to those of 2,4-pyridinedicarboxylato triaqua cadmium(II) hemihydrate, Co
2+, and Cd
2+ containing pyrazinamide, reported elsewhere [
21,
26,
27,
28].
3.1. Description of the Crystal Structure of [Mn(2,6-pydc)2](imi) (1) and [Co(H2pza)2(H2O)2(NO3)]•NO3 (2)
The crystal data collection and structure refinement parameters for the cocrystals and crystals of compounds
1 and
2 are highlighted in
Table S5.
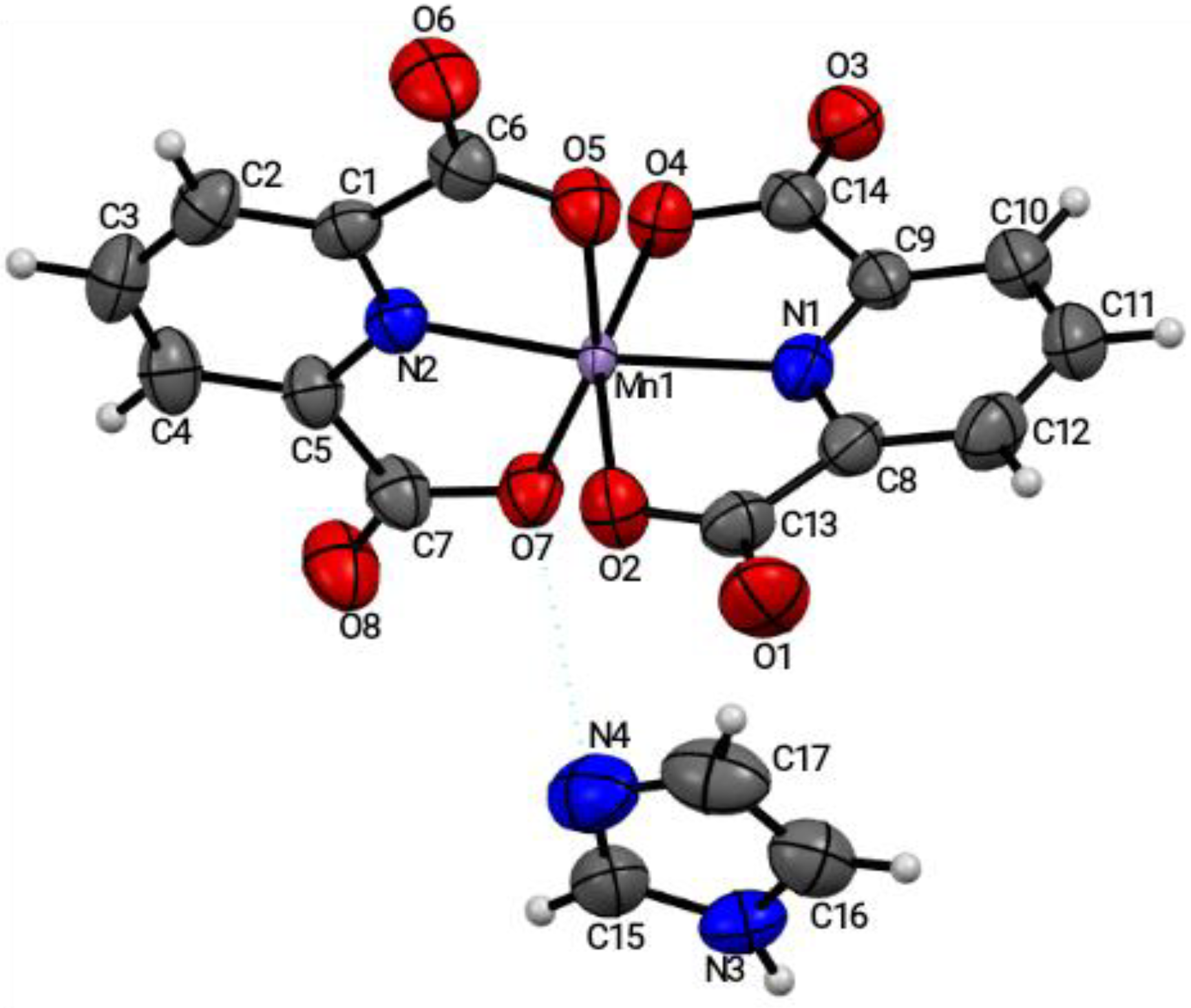
[Mn(2,6-pydc)
2](imi)
1 crystallized in the orthorhombic Pbca space group, with the asymmetric unit consisting of one Mn(II) atom bound by two 2,6-pydc molecules, and one molecule of imidazole lying outside the coordination sphere (see
Figure 1). The central Mn(II) atom lay in an undistorted octahedral coordination environment with an N
2O
4 donor set; the adjacently-coordinated 2,6-pydc ligands, containing O2, O4, O5, and O7 atoms, were constructed in such a way that both occupied the axial and equatorial planes. Each of the 2,6-pydc carboxylate groups exhibited the µ
1–ղ
1:ղ
0-monodendate coordination mode [
22,
27]. The [Mn(COO)
2] structural building unit (SBU) could be considered as a 2-connected node, in which the Mn(1) sat on an inversion center, and was defined by four carboxylate oxygen atoms, which formed a finite two-dimensional (2D) structure.
In addition, the uncoordinated imidazole molecule, situated at the equatorial plane outside the coordination sphere, was linked to the framework by hydrogen bond interactions via N4 and N3 (calc. distance = 3.012 Å). The hydrogen bond and/or van der Waals’ interactions between the uncoordinated imidazole and the coordinated 2,6-pydc were labeled as N3⸺H3a⋯O3 and O7⋯N4; these bonds stabilized the two-dimensional (2D) structure of compound
1 (see
Figure S7). It was also evident that O7⋯N4 constituted the van der Waals’ contact. The hydrogen bond parameters for [Mn(2,6-pydc)
2](imi) are shown in
Table S6. Furthermore, the position of the Mn1–O2 (2.000 Å) and Mn1–O4 (2.011 Å) bonds, of one 2,6-pydc
- moiety, were in a normal range with those of the adjacent Mn1–O5 (2.023 Å) and Mn–O7 (1.998 Å) bonds, indicating that the assigned Mn-O bond energies were perpetually the same (
Figure S3). Similarly, the O2–Mn1–O4 (157.90°) angle of one 2,6-pydc
- moiety occupying an axial position deviated by a small angle of ca. 0.21° from the O5–Mn1–O7 (157.69°) of the adjacent 2,6-pydc moiety, showing that the angles were in plane, and that there was bite distortion from the linearity [
27]. Distances of 1.957 Å and 1.971 Å between the (Mn1-N1) and (Mn1-N2) bond were shorter than the Mn1–O bonds (bond range was from 1.998 Å to 2.023 Å), indicating that the bond energies, E
b between the Mn
2+–N atoms (of 2,6-pydc), were stronger than the Mn
2+–O atoms of 2,6-pydc [
29]. The selected bond distance and angles of compound
1 are presented in
Table S7.
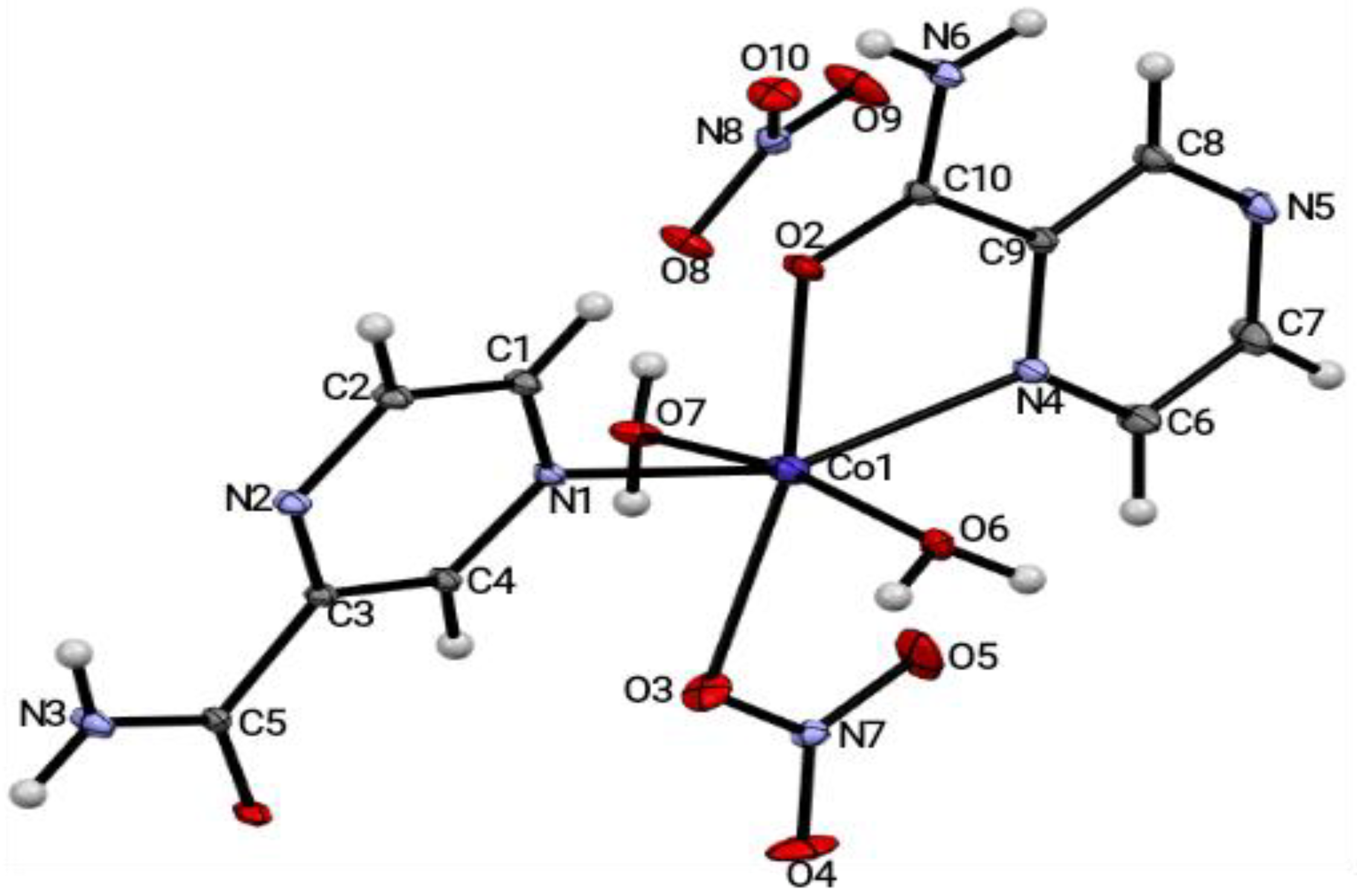
The crystal structure of [Co(H
2pza)
2(H
2O)
2(NO
3)]•NO
3 (
2) in
Figure 2 crystallized in the triclinic P-1 space group, with one Co(II) metal atom, two pyrazinamide (H
2pza) molecules, two water molecules, and two nitrate molecules in the asymmetric unit. The crystal structure revealed also that the cobalt ion was being coordinated by: (i) two H
2pza molecules acting as the unidendate donor ligand; (ii) two water molecules; and (iii) one nitrate molecule within the coordination sphere, which provided the counter ion for the Co
2+ atom; the uncoordinated nitrate molecule also provided the counter ion to enhance the rigidity (stability) of the framework. The resulting oxidation state of the cobalt ion in the crystal structure was +2 [
25].
The average bond length of Co1-N was 1.8070 Å, while that of Co1-O was 2.1968 Å, respectively. This observation was similar to the average bond lengths of Co-N and Co-O found for copper (II) bound by pyrazinamide and 2-nitrobenzoate [
24]. The bond angles of O-Co1-N ranged from 80.26(5)° to 125.44(5)°, while O-Co1-O ranged from 84.25(5)° to 97.57(6)°, respectively. The average bond length of Co-O (2.1968 Å) suggested that the bond energies (E
b) between Co-O were weaker than those of Co-N [
21,
30]. Selected bond lengths and bond angles in compound
2 are highlighted in
Table S8.
The distance, 4.658 Å, between the Co1 atom and the N8 atom of the uncoordinated nitrate molecule was within the range reported for other coordination polymers [
25]. The coordinated water molecules were bound to the Co
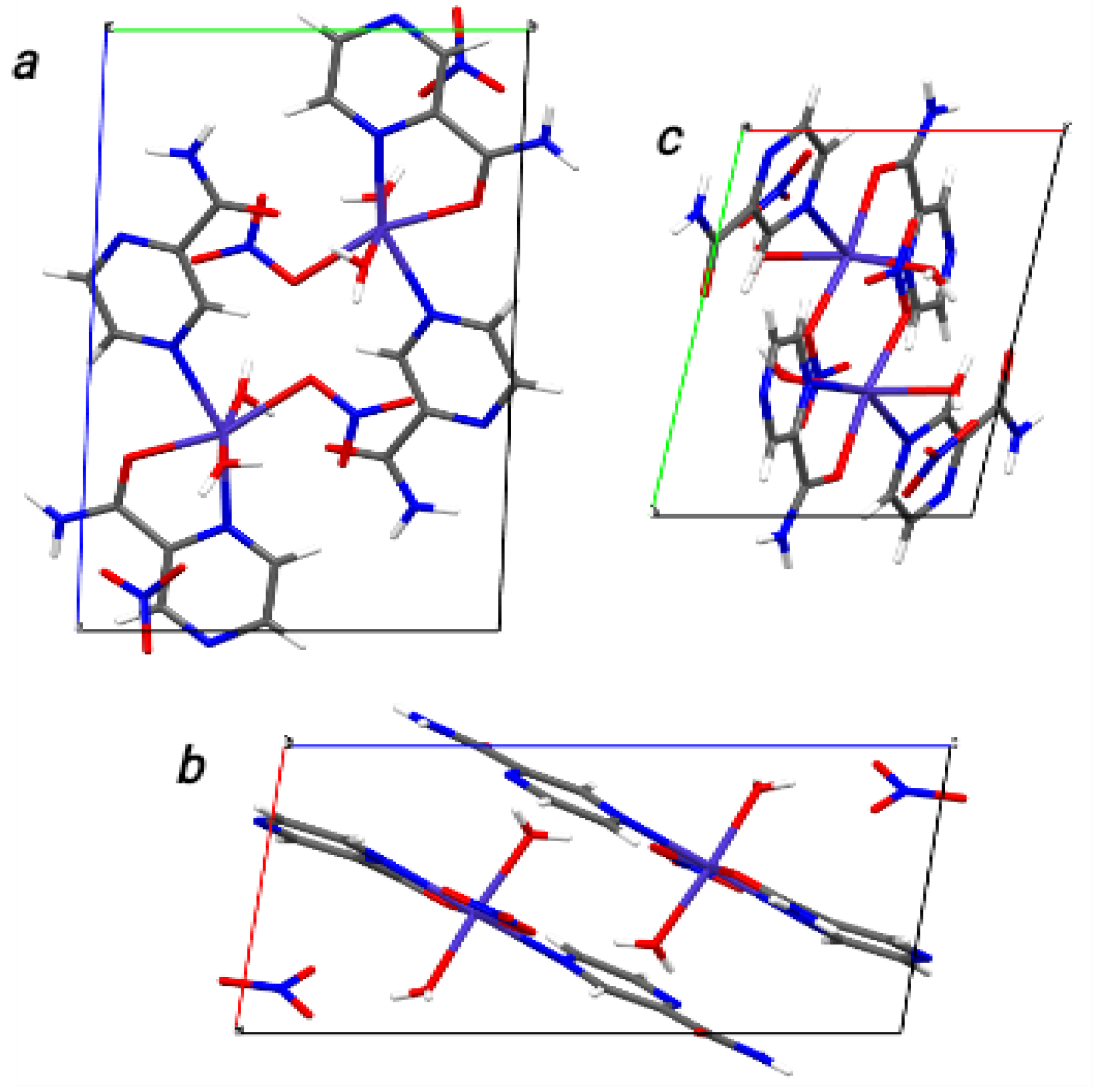
2+ ion by ionic bonds, and formed pendants for the 1D structure along the crystallographic b plane, as shown in
Figure 3.
The 1D structure showing hydrogen bonds in
Figure S8 indicates that the free nitrate and coordinated water molecules formed a network of hydrogen bonds, in which the linkers (hydrogen bonds) ran along the
c-axis in the direction of the coordinated nitrate, and along the
a-axis in the direction of the free nitrate and coordinated water molecules [
25]. Furthermore, the intra-molecular hydrogen bond interactions in compound
2 involved those of aromatic amine, carbonyl groups, coordinated nitrate, O6W, and O7W, and they were labeled as (O6–H6C⋯O1), (O6–H6D⋯O3), (O7–H7A⋯O1), and (N3–H3B⋯N5), respectively. The inter-molecular hydrogen bond interactions involved those of aromatic amine groups, O7W, and free nitrate, and they were labeled as (O7–H7B⋯O8), (N3–H3A⋯O10), and (N6–H6B⋯O9). The parameters for the intra- and inter-molecular hydrogen bond interactions are summarized in
Table S9.
3.2. Powder X-ray Diffraction Patterns (PXRD) of [Mn(2,6-pydc)2](imi) (1) and [Co(H2pza)2(H2O)2(NO3)]•NO3 (2)
Powder XRD patterns (Cu, Kα) of compounds
1 and
2 (
Figures S9 and S10) were conducted, to probe the characteristic phases related to the active sites in both materials prior to their usage as catalysts. Both samples were activated at 100 °C prior to catalytic application. However, the simulated PXRD patterns of compounds
1 and
2 displayed characteristic diffraction phases, with very slight variation where peaks disappeared for the as-synthesized patterns. The simulated patterns of compounds
1 and
2 displayed peaks at 2
Ө = [11.4°, 12.2°, 12.9°, 14.2°, 22.9°, 23.7°, 26.4° and 30.4°], and 2
Ө = [6.3°, 11.9°, 13.3°, 15.5°, 16.4°, and 23.6°], which corresponded to [(100), (004), (102), (112), (200), (211), (222), and (233)] and [(001), (011), (100), (101), (012), and (112)] planes; these were typical of MnO and CoO accessible sites, respectively. Furthermore, it was observed that the PXRD patterns of compunds
1 and
2 in crystalline powder form were consistent with the simulated pattern in single-crystal form; this implied that the structural integrity of both materials was retained after crushing to powder form [
31].
3.3. High Resolution (HR) TEM Images of 1 and 2
The TEM images of compound
1 scanned at 100 nm and 0.2 µm revealed flake-shaped morphology consisting of pore walls attributed to crystallites co-assembling to form a moderate ordered mesostructure. The TEM images of compound
2 were scanned at 20 nm and 100 nm, and gave block-shaped morphology; the inset (selected area electron diffraction, SAED) illustrates that crystallinity (spots) and Bragg’s reflection were from an individual crystallite [
22]. The HRTEM images of compounds
1 and
2 are shown in
Figure S11A,B, respectively.
4. Oxidation of Cyclohexanone
The strong pre-adsorption of cyclohexanone on a catalyst’s surface is an important phenomenon for an efficient chemical transformation to take place, as it affects the rate and mechanism of transformation [
2,
9]. For this reason, before examining the catalytic activity of each compound, thermal stability tests were performed, using the TG analytical technique. In addition, thermal activation of the samples was carried out at 100 °C under vacuum in air. Thermal activation of each sample was done to: (i) remove lattice water and other uncoordinated molecules; and (ii) generate the coordinatively unsaturated metal sites (acidic sites). The typical reaction scheme for the catalytic process is illustrated in
Scheme 3.
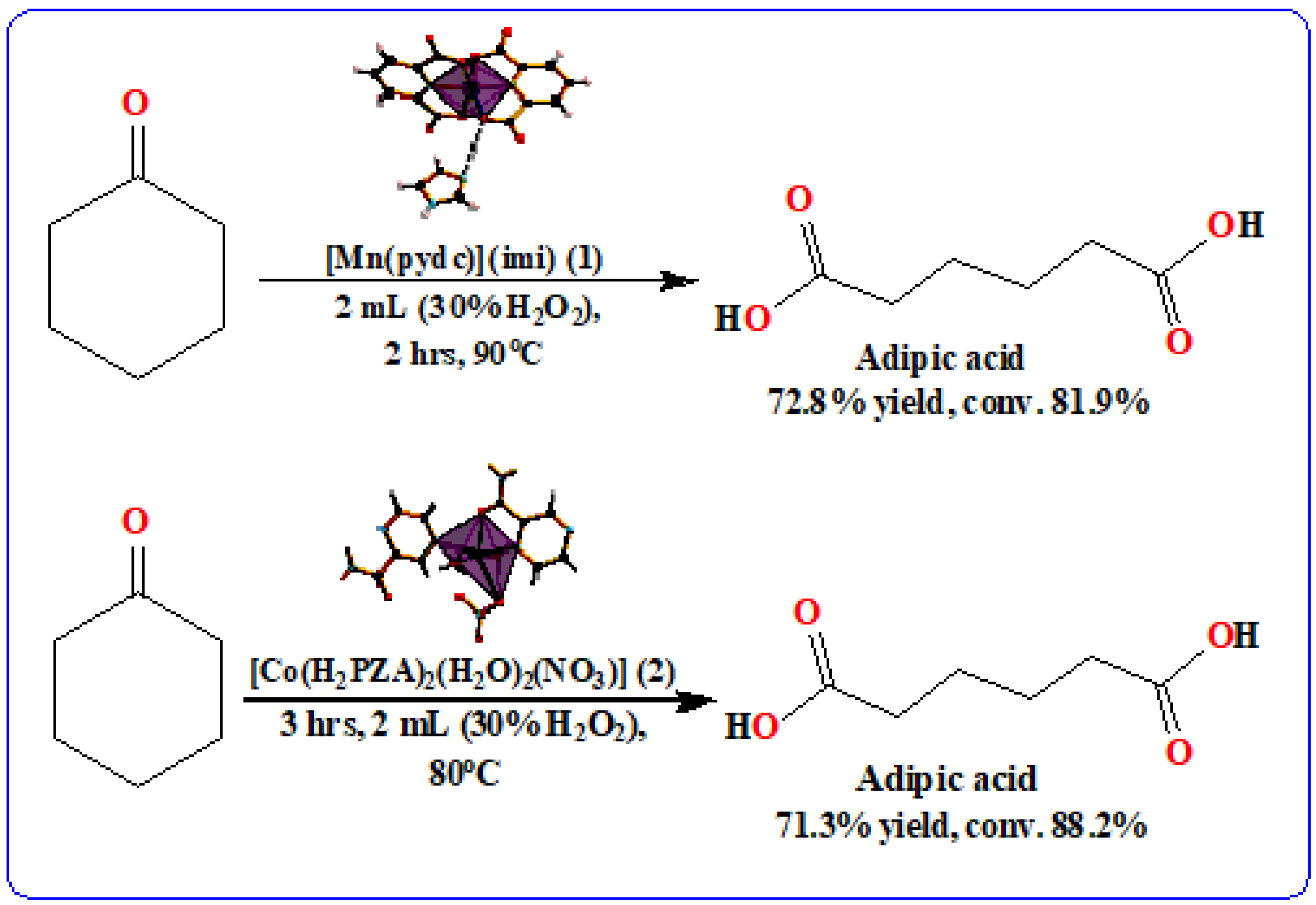
Compounds
1 and
2, with different ligands, were able to catalyze each reaction, such that the +2 oxidation states of the metals were retained. The use of one or two ligands provided high selectivity to AA. The isolated yields of AA, as shown in
Scheme 3, indicated that the use of the Mn-coordinated compound (
1) gave a 72.8% yield of AA, while compound
2, acting as Co
2+ catalyst, gave a 71.3% yield of AA. The side products obtained gave very low yields, such as 6-hydroxyhexanoic acid and ε-caprolactone. Furthermore, it was observed that the thermal activation of each candidate material enhanced their hydrolytic stability, and favored the catalytic activity of both compounds. The oxidation of cyclohexanone was also promoted with the use of 30% H
2O
2. The selected catalytic activity of both complexes under study is summarized in
Table 2 below.
Control experiments were performed at 30 °C and at temperatures above 100 °C, giving low yields, (21.0% and 36.6%), of AA. This suggests less activation energy input at 30 °C and poor performance catalysts at temperatures above 100 °C due to defects in active sites. By observation, an appreciable yield of AA was obtained over the Mn(II) and Co(II) catalysts (
1), on the basis that there was a complete charge balancing by the acidic ligands, and formation of a water soluble complex which made contact with an oil phase (cyclohexanone) [
2]. It was suspected also that the conversion of cyclohexaonne to AA was favored by acetonitrile, which prevented water from accessing the active sites of both catalysts; this enhanced mass transfer, and resulted in an accelerated reaction rate [
32]. The catalysts also appeared to be retained within the solution through multiple cycles of re-use, and were not dissolved in water; in other words, the structural integrity of both complexes was retained. The effects of other factors on the reaction were studied as follows.
4.1. Effect of Reaction Temperature
The experiment was initially performed at low temperature, 40 °C, giving low conversions of cyclohexanone [
Table 2, Entries 2 and 7]. This was attributed to the limited catalytic activity of compounds
1 and
2 at low temperatures. The isolated yields of AA increased as the reaction temperature was increased gradually, by observation. Optimum yields of AA (72.8% and 71.3%) were recorded at 80 °C [
Table 2, Entries 5 and 9]; this suggests the practical applicability of Mn(II)- and Co(II)-coordinated compounds to accelerate the direct conversions of cyclohexanone (81.9% and 88.2%). The selectivity of AA was 64.1% and 67.2%, respectively, in the presence of H
2O
2 at 80 °C. Furthermore, the use of deionized (DI) water, i.e., 41.6% at 40 °C, increased the liquid yield of caprolactone peroxide, while the measurement of 20.8% of DI favored the target product yields [
Table 2, Entries 2 and 7] [
4,
33]. The TOF of the oxidation reactions at different reaction temperatures was 40 °C (6.4 h
−1 @
1 and 1.4 h
−1 @
2) < 50 °C (14.7 h
−1 @
1 and 4.9 h
−1 @
2) < 60 °C (16.3 h
−1 @
1 and 5.4 h
−1 @
2) < 70 °C (22.7 h
−1 @
1 and 7.3 h
−1 @
2) < 80 °C (28.7 h
−1 @
1 and 18.5 h
−1 @
2), respectively. The graphical illustration for the effect of the reaction temperature is shown in
Figure S12.
4.2. Effect of Catalyst Loading
As varying degrees of temperature affect the performance of a catalyst, the amounts of catalysts loaded into the catalytic systems were studied. In one of the experiments, the absence of catalysts gave only a traceable amount of AA over compounds
1 and
2 [
Table 2, Entries 1 and 6]; this indicated the dependence of the reaction on active species present in the catalytic system, i.e., Mn
2+ and Co
2+ ions [
29]. An HPLC chromatogram of a product (traceable amount of AA, 2µL), taken and analyzed in the absence of catalyst
1, has been shown in
Figure S13. The use of Mn(NO
3)
2.4H
2O (MnN) and Co(NO
3)
2.4H
2O (CoN) were also examined; these gave very low yields of AA (12.5% over compound
1, and 22.1% over compound
2) [
Table 2, Entries 10 and 11]. However, the oxidation of cyclohexanone over low amounts of compounds
1 and
2 (1.4 × 10
−4 mol L
−1) gave moderate yields: 64.0% and 48.7% of AA [
Table 2, Entries 3 and 8]. There were improved product yields, 72.8% and 88.2%, when the amounts of catalysts were slightly increased [
Table 2, Entries 5 and 9]. This also suggests the dependence of the reaction on more Lewis acidity coverage with respect to the bulk solution, thereby enhancing the inter-cavity diffusion of cyclohexanone through the catalysts’ pore walls [
26,
34,
35]. The TOF of the oxidation reactions of different loading was 0.00074 mol L
−1 (6.4 h
−1 @
1) < 0.0014 mol L
−1 (14.8 h
−1 @
1) < 0.0022 mol L
−1 (19.2 h
−1 @
1) < 0.0029 mol L
−1 (28.7 h
−1 @
1) and 0.00071 (1.4 h
−1 @
2) < 0.0014 mol L
−1 (3.8 h
−1 @
2) < 0.0021 mol L
−1 (18.5 h
−1 @
2), respectively. An illustration of the effect of catalyst loading on the oxidation of cyclohexanone is presented in
Figure S14.
4.3. Effect of Reaction Time
An experiment was conducted at the reaction time of 40 min on compounds
1 and
2, which gave 17.8% and 22.7% yields of AA [
Table 2, Entries 12 and 14]. As the reaction progressed by gradually increasing the time, the full catalytic potentials of compounds
1 and
2 were released, giving optimum conversions/selectivity of AA (81.9%/64.1% and 88.2%/67.2%) after 2 and 3 hrs, respectively, [
Table 2, Entries 5 and 9]. The TOF of the oxidation reaction at variable times was 40 min. (6.1 h
−1 @
1 and 6.9 h
−1 @
2) < 120 min (28.7 h
−1 @
1) < 180 min (18.5 h
−1 @
2), respectively. The graphical illustrations for the conversion of cyclohexanone at different reaction times are shown in
Figure S15A,B. The HPLC chromatogram of AA attributed to the maximum activity of compound
1 on the reaction system is shown in
Figure S16. These results are consistent with those reported elsewhere [
3,
36].
4.4. Effect of Oxidant
The effect of oxidant on the catalytic performance of compounds
1 and
2 was monitored by reacting mixtures in the absence of oxidant, H
2O
2. The quantified amounts of AA were not promoted by this attempt. However, 53.7% and 65.2% conversions of cyclohexanone were recorded with 85.2 mmol of H
2O
2 [
Table 2, Entries 13 and 15]. Optimum yields of AA, 72.8%, and 71.3%, were also recorded with the use of 170.4 mmol of H
2O
2 after 2 and 3 hrs of reaction time over compounds
1 and
2, respectively, [
Table 2, Entries 5 and 9].
Tella, et al. [
32], reported the formation of by-product of water molecules during the decomposition of H
2O
2, which could be attributed to the conversions, 53.7% and 65.2%, recorded with H
2O
2, 85.2 mmol.
Furthermore, investigation of UV-visible spectral analysis of reactive systems containing compounds
1,
2, and oxidant (H
2O
2) were carried out in the presence and absence of cyclohexanone. Studies showed that the reactive species, Mn and Co, gave characteristic absorption bands in the absence of cyclohexanone, at a constant temperature of 80 °C at 240 nm (π → π*), 370 nm (n → π*), and 512 nm (d-d transition) for compound
1; and 264 nm (π → π*), 361 nm (n → π*), and 722 nm (d-d transition) for compound
2 (
Figure S17, 1a and 2a); this was attributed to Mn(II)-H
2O
2 and Co(II)-H
2O
2 adducts formation. Reduced concentrations of compounds
1 and
2 were traceable to the addition of cyclohexanone, leading to a slight decrease of spectral intensity at 512 nm for compound
1 (
Figure S17; 1b), and the disappearance of spectral band, coupled with a slight shift of the spectral band from the initial 722 nm to 734 nm for compound
2 (
Figure S17; 2b). This was attributed to the formation of Mn-H
2O
2-cyclohexanone and Co-H
2O
2-cyclohexanone intermediates or adducts, due to reactive species oxidization from Mn(II) and/or Co(II) to Mn(III/II) and/or Co(III) in H
2O
2 medium via the enhanced charge transfer mechanism, before completing the catalytic process [
32,
36].
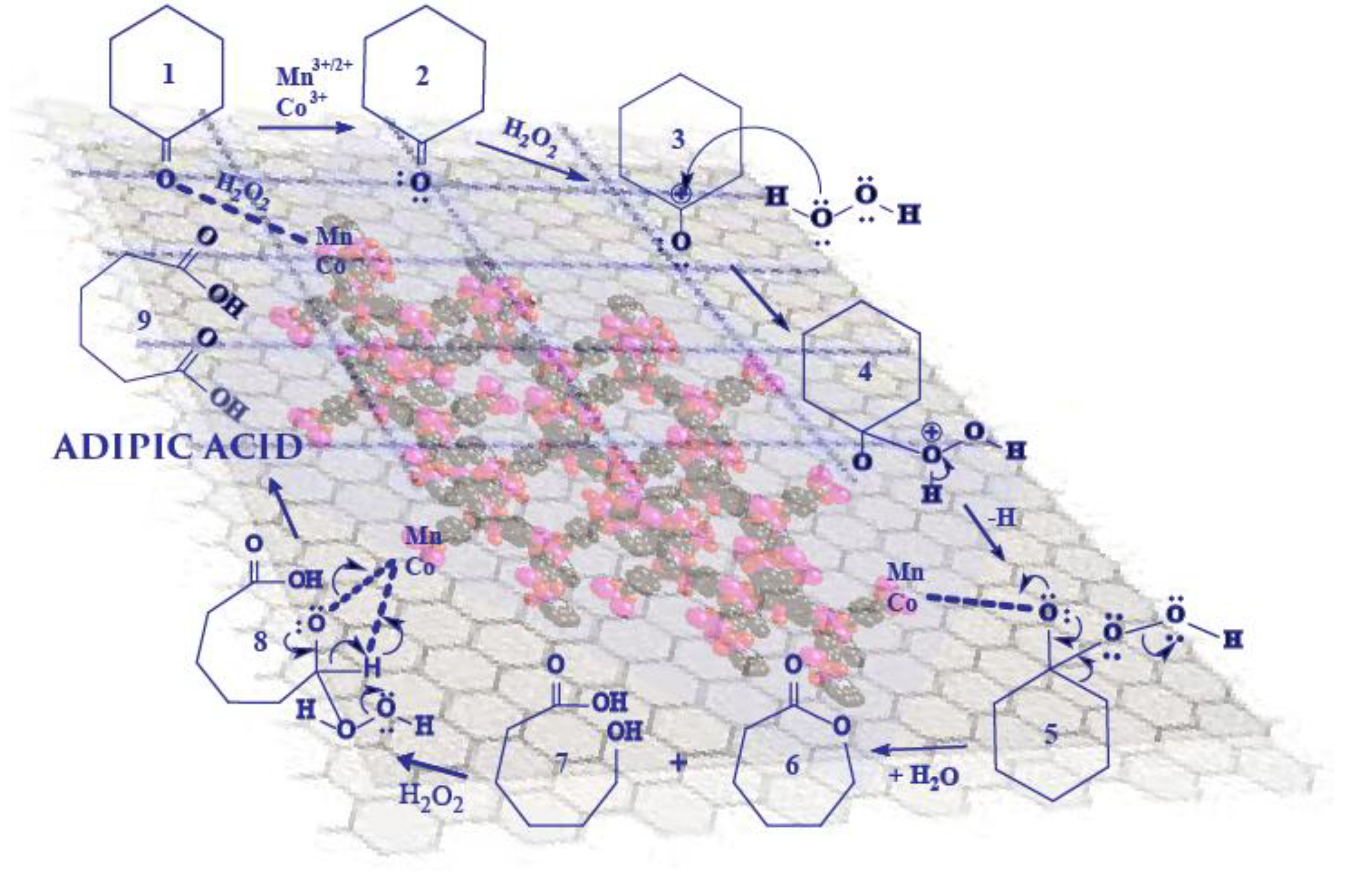
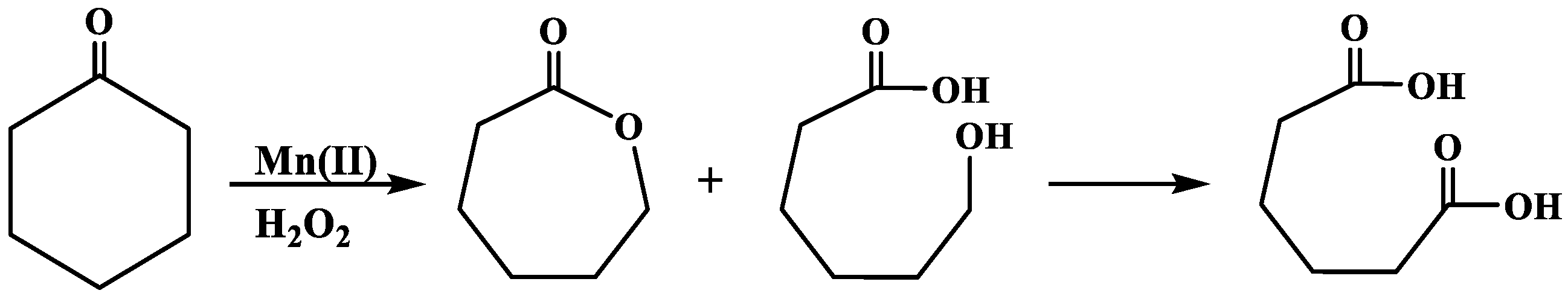
Xia, et al. [
37] also reported a mechanism called Baeyer–Villiger oxidation, which has been used to describe the oxidation of cyclohexanone with H
2O
2 in different systems. The modified reaction mechanism is comprised of steps 1 to 9, as shown in
Scheme 4 below.
Mn(II) and Co(II) was first activated via enhanced oxidation in an H
2O
2 medium, to form active Mn(III/II)–H
2O
2 and Co(III)–H
2O
2 adducts (steps 1 and 2) [
32]. Charge transfer occurring from Mn-H
2O
2 and Co-H
2O
2 resulted in the diffusion of cyclohexanone onto an active surface (catalyst) to form reactive Mn-H
2O
2–cyclohexanone and Co-H
2O
2–cyclohexanone intermediates. The adsorbed cyclohexanone underwent rearrangement and/or breaking of bonds (steps 3 and 4), followed by decomposition of H
2O
2 in step 4, which formed OH
• and CyOO
• radicals. Deprotonation of a hydrogen ion and desorption reaction in step 5 led to the formation of ε-caprolactone, and water as a byproduct of the decomposition of the H
2O
2. Further desorption reaction led to the formation of 6-hydroxyhexanoic acid in step 6, while continual activation of 6-hydroxyhexanoic acid in the H
2O
2 medium (steps 7 and 8) favored the formation of AA in step 9. The quantification of the reaction products was in good agreement with those reported elsewhere [
2,
19]. The TOF of the reaction with H
2O
2 was 85.1 mmol (18.8 h
−1 @
1 and 15.2 h
−1 @
2) < 170.4 mmol (28.7 h
−1 @
1 and 18.5 h
−1 @
2), respectively. The illustration for the formation of the two intermediates is as shown in
Scheme 5.
The target product (2 µL) was authenticated by conducting
1H NMR and
13C NMR spectroscopic analyses. The
1H NMR signal (
Figure S18) at optimum reaction conditions revealed chemical shifts at 3.26–3.70 δ (ppm), and was assigned to methylene protons–CO–(CH
2)
n–CO– of AA; this was denoted as ‘b’ and ‘c’. The signal for aliphatic protons attached to carboxylate groups (C=O–OH) appeared at 11.96 δ (ppm), and was denoted as ‘a’.
Similarly, the
13C NMR spectrum shown in
Figure S19 revealed two sharp signals at 20.34 δ (ppm) and 34.27 δ (ppm), which were assigned to methylene carbon atoms, (–CH
2)
n. These were also denoted as ‘b’ and ‘c’. The carbon atoms attached to the carboxylate group displayed signals at 179.82 δ (ppm), and were denoted as ‘a’. Both the
1H and
13C NMR spectra reported herein were comparable to those reported in the literature [
23].
Jianhui, et al. [
12], compared the product selectivity (mol %) of KA oil in oxidation with H
2O
2 over metal catalysts, as a basis for recyclability evaluation; the result of our studies indicated that the isolated yield in adipic acid was maintained after the first and second cycle of reuse. A loss in catalytic activity was observed after the third cycle of reuse, due to a coordinative mismatch between the metal nodes and linkers, and the inherent Lewis acidity defects (
Figure S20) [
14].
MP-AES analysis was further carried out to ascertain the active metal contents in both compounds (
Table 3). The results gave low Mn
2+ and Co
2+ contents in compounds
1 and
2, suggesting a reduced number of coordinatively unsaturated sites (CUM) [
14,
28].
5. Kinetics and Thermodynamics of Cyclohexanone Oxidation
The kinetics of cyclohexanone-catalyzed reactions were studied by following a theoretical study by Kumal, et al., 2008 [
38]. The rate law, using the Langmuir–Hinshelwood (L-H) mechanism, of the unimolecular reaction was expressed as:
where
r = rate of reaction, [
A] = final concentration of cyclohexanone,
k1 = adsorption rate constant,
k−1 = desorption rate constant, and
k2 = decomposition rate constant. The slope and intercept corresponded to the above rate constants, and was obtained in an L-H linear plot of 1/
r vs. 1/[
A]. The L-H plot obtained from this study is presented in
Figure S21.
The L-H plot parameters obtained for the oxidation of cyclohexanone are summarized in
Table 4 below:
In
Table 4, it is evident that the regression coefficient values, R
2, suit the L-H model used for describing the adsorption process [
38,
39]. The positive
k1 (adsorption constant) values indicate favorable monolayer adsorption of cyclohexanone at the unsaturated active sites. The negative equilibrium (
k−1) values suggest re-distribution of free energy in the catalytic systems at equilibrium, while the positive
k2 values (desorption constants) suggest high rates of decomposition and diffusion of AA from an active surface [
39]. Also, the observed
k2 >>
k1 signified monolayer relaxation and reconstruction of the active surface during adsorption, and aggregation of the substrate (cyclohexanone), which has low interference with binding sites; this was further attributed to enhanced (fast) dissociation of adipic acid from the active surface. In addition, the rate of reaction,
r (Ms
−1), was faster over compound
1 than over compound
2.
6. Thermodynamic Parameters for the Oxidation of Cyclohexanone
The thermodynamic parameters obtained with compounds
1 and
2 are presented in
Table 5. The activation entropy values, ∆
S‡, and the enthalpy of the transition state, ∆
H‡, were determined by using the Arrhenius and Eyring equations [
39,
40,
41]:
where
k1 = initial rate constant,
kB = Boltzmann’s constant (1.3 × 10
−23 J.K
−1),
T = temperature in Kelvin, and
h = Plank’s constant (6.626 × 10
−34 J.s). These values, ∆
S‡ and ∆
H‡, are recorded in
Table 3 and
Table 4, and were obtained in a linear plot (y = mx + c) of ln (
k1/
T) vs. 1/
T, as shown in
Figure S22. The intercept corresponded to ln
kB/
h + ∆
S‡/
R; the slope corresponded to ∆
H‡/
R. The Gibb’s free energy value, ∆
G‡, was obtained from the expression: ∆
G‡ = ∆
H‡ −
T∆
S‡. The activation energy, E
a, was calculated using the expression: E
a = ∆
H‡ +
RT, where
R = universal gas constant (8.314 J mol
−1 K
−1).
As shown in
Table 5, the ∆
G‡ values (−37,256 J mol
−1 and −38,508 J mol
−1) suggest an initial reactivity of non-thermalized molecules at low temperatures [
39,
41]. However, as the temperature of the catalytic system gradually increased, free energy was distributed, leading to an increase in the negative ∆
G‡ values (−43,206 J mol
−1, and −43,428 J mol
−1), via a spontaneous catalytic process. This further indicates that the physisorption condition, under which the adsorption of the cyclohexanone took place on an active surface, became more negative and more favorable by increasing the reaction temperature [
40]. The positive entropy, ∆
S‡ values (119.0 JK
−1, and 122.6 JK
−1), suggested that the reacting molecules were liberated from their initial state, to a transition state, due to an increase in the degrees of freedom (∆
S‡) [
39]. Though a transition state was reached which promoted radical reactions, as earlier described in the mechanism of reaction, the radical reactions (of hydroxyl, OH
•, and carboxyl, CyOO
•, groups) led to pre-formation of complex or intermediate products (6-hydroxyhexanoic acid and caprolactone). The Arrhenius and Eyring plots of cyclohexanone oxidation over compounds
1 and
2 are presented in
Figure S22. Furthermore, the positive enthalpy, ∆
H‡, values suggest that at optimum temperature, the equilibrium of the catalytic systems moved in the direction that tended to promote complete AA formation [
39]. Similarly, as the enthalpies, ∆H
‡, of the adsorbed substrate (2.79 KJ mol
−1, and 2.81 KJ mol
−1) over compounds
1 and
2, were less than 40 KJ mol
−1, this indicated that physisorption primarily occurred at the coordinatively unsaturated metal (CUM) sites, and that the host-guest interactions involved those of van der Waal’s interactions or weak π-π interactions [
39]. The partial concave curve (
Figure S22) observed for compounds
1 and
2 was attributed to two rate-limiting reaction steps. In addition, the activation energy, E
a value (3021 J mol
−1), suggested that compound
1 required more activation energy to catalyze the oxidation of cyclohexanone than compound
2 (2938 J mol
−1), which required less activation energy. The use of acetonitrile as solvent suggested that the access of water into the pore walls of the catalysts was prevented; hence, the reaction rate was enhanced. The TOF of the catalytic reaction with acetonitrile gave 22.9 h
−1 and 13.9 h
−1, respectively.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}