







Mono-Alkyl-Substituted Phosphinoboranes (HRP–BH2–NMe3) as Precursors for Poly(alkylphosphinoborane)s: Improved Synthesis and Comparative Study


Abstract
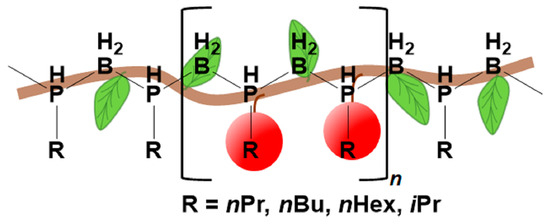
:
1. Introduction
2. Results and Discussion
2.1. General Synthetic Procedure
2.2. Phosphinoboranes with Primary Alkyl Substituents
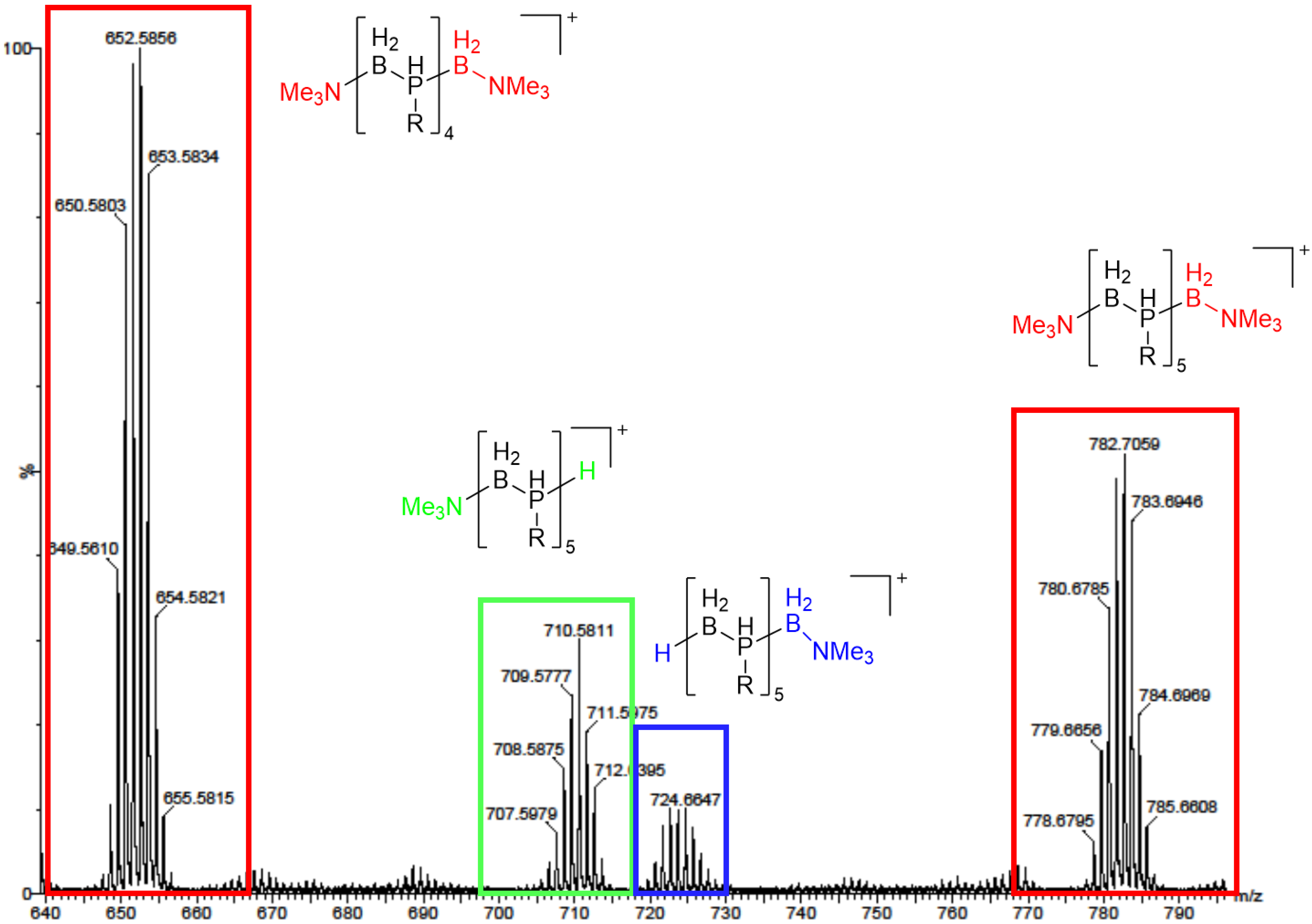
Preliminary Investigations of 1a–c as Polymer Precursors
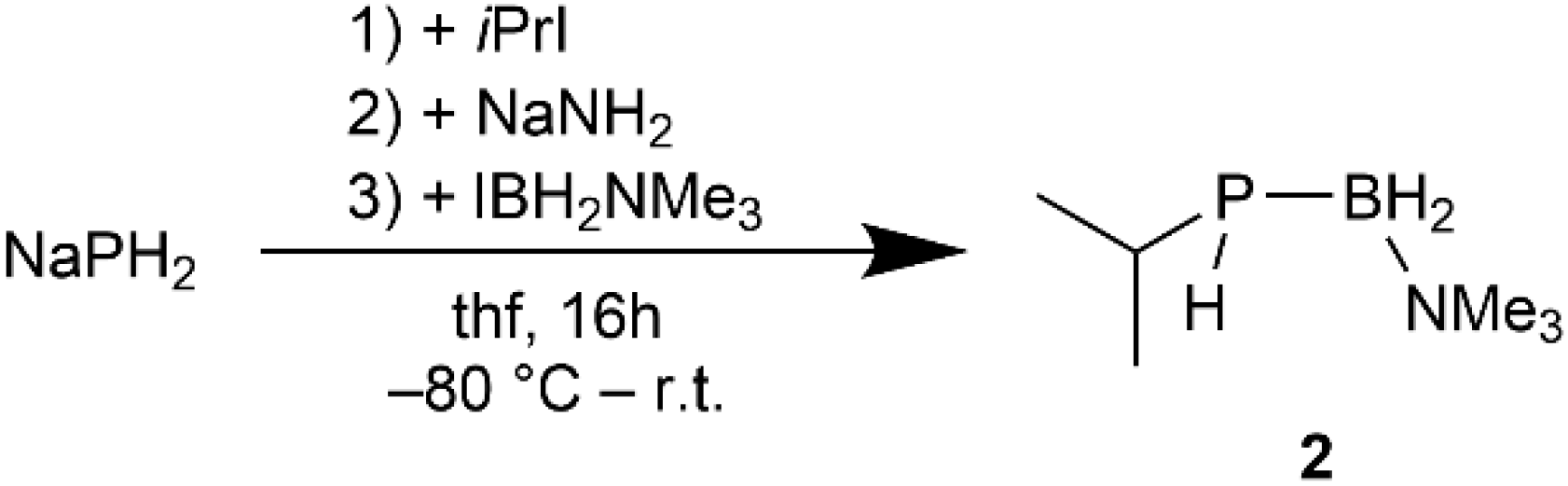
2.3. Phosphinoboranes with Secondary Alkyl Substituents
2.4. Phosphinoboranes with Functionalized Alkyl Substituents
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of nPrPHBH2NMe3 (1a), nBuPHBH2NMe3 (1b), and nHexPHBH2NMe3 (1c) by Adjusted Literature Procedures
- 1a: 72 mg (0.49 mmol, 28%).
- 1b: 57 mg (0.46 mmol, 13%).
- 1c: 125 mg (0.67 mmol, 38%).
3.3. One-Pot Synthesis of nPrPHBH2NMe3 (1a), nBuPHBH2NMe3 (1b), nHexPHBH2NMe3 (1c)
- 1a: m = 0.414 g (2.82 mmol, 29%)
- 1b: m = 1.537 g (9.5 mmol, 54%)
- 1c: m = 1.256 g (6.6 mmol, 66%)
3.4. Synthesis of [iPrPH2BH2NMe3]AlCl3I
3.5. Synthesis of iPrPHBH2NMe3 (2)
3.5.1. From [iPrPHBH2NMe3]AlCl3I
3.5.2. From a One Pot Synthesis
3.6. Synthesis of Me3NBH2PH2C4H8PH2BH2NMe3 (3a) and H2PC4H8PH2BH2NMe3 (3b)
3.7. Polymerization Experiments
3.7.1. Of 1a
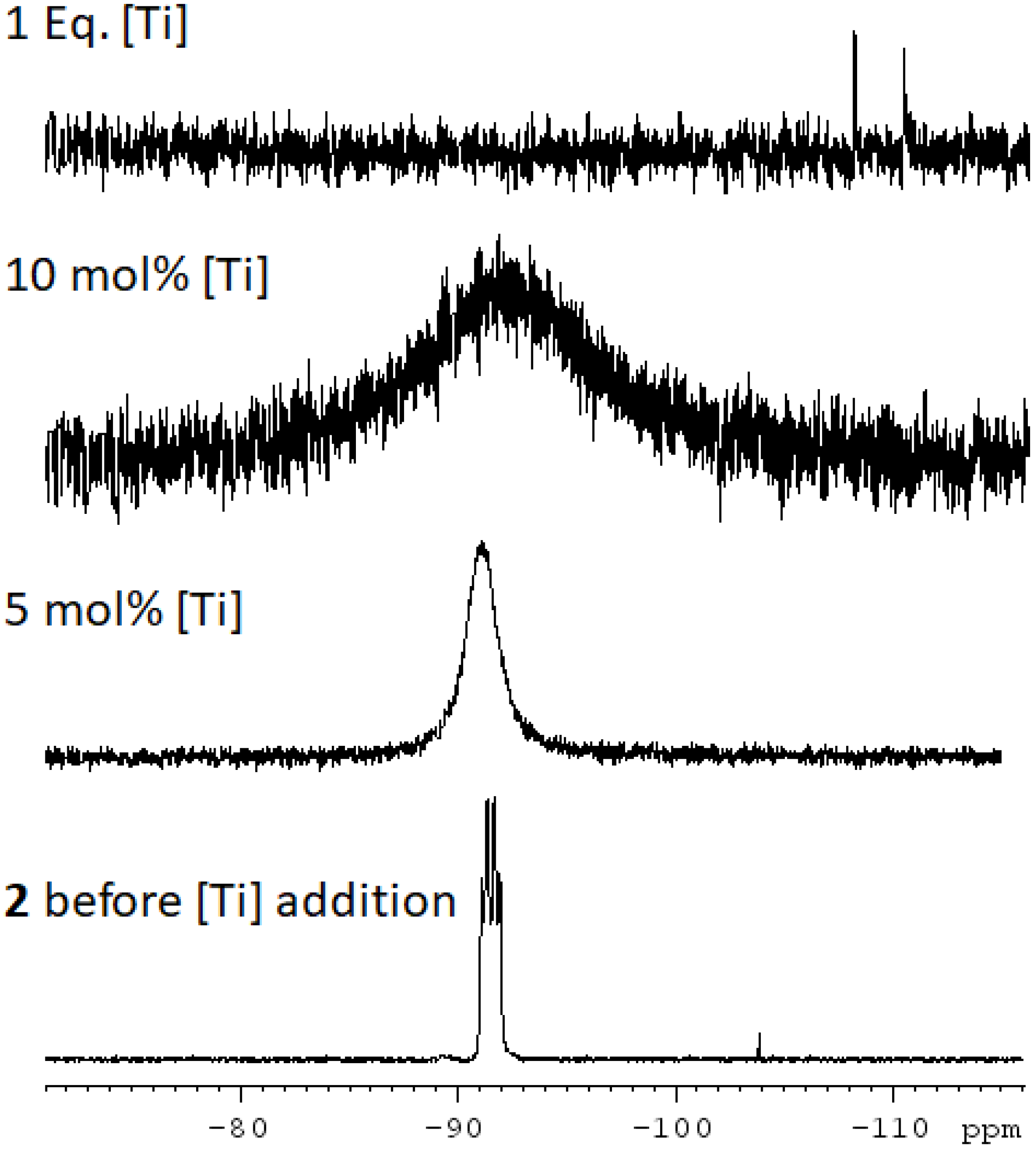
3.7.2. Of 1b under Catalytic Conditions
3.7.3. Of 1c
3.7.4. Of 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chanda, M.; Roy, S.K. Industrial Polymers, Specialty Polymers, and Their Applications; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Aguilar, M.R.; Román, J.S. Smart Polymers and Their Applications; Woodhead Publishing: Sawston, UK, 2019; pp. 1–11. [Google Scholar]
- Gauthier, M.A.; Gibson, M.I.; Klok, H.A. Synthesis of functional polymers by post-polymerization modification. Angew. Chem. Int. Ed. 2009, 48, 48–58. [Google Scholar] [CrossRef]
- Carraher, C.E.; Moore, J.A. Modification of Polymers; Springer Science & Business Media: Berlin, Germany, 2012; Volume 21, pp. 1–24. [Google Scholar]
- Liang, M.; Manners, I. Poly(thionylphosphazenes): A new class of inorganic polymers with skeletal phosphorus, nitrogen and sulfur (VI) atoms. J. Am. Chem. Soc. 1991, 113, 4044–4045. [Google Scholar] [CrossRef]
- Allcock, H.R. Crosslinking reactions for the conversion of polyphosphazenes into useful materials. Chem. Mater. 1994, 6, 1476–1491. [Google Scholar] [CrossRef]
- Honeyman, C.H.; Manners, I.; Morrissey, C.T.; Allcock, H.R. Ambient temperature synthesis of poly(dicholrophosphazene) with molecular weight control. J. Am. Chem. Soc. 1995, 117, 7035–7036. [Google Scholar] [CrossRef]
- Archer, R.D. Inorganic and Organometallic Polymers; Wiley VCH: New York, NY, USA, 2001; pp. 179–226. [Google Scholar]
- Manners, I. Polymers and the periodic table: Recent developments in inorganic polymer science. Angew. Chem. Int. Ed. 1996, 35, 1602–1621. [Google Scholar] [CrossRef]
- Clarson, S.J.; Semlyen, J.A. Siloxane Polymers; Prentice Hall: Upper Saddle River, NJ, USA, 1993; pp. 616–648. [Google Scholar]
- Neilson, R.H.; Wisian-Neilson, P. Poly(alkyl/arylphosphazenes) and their precursors. Chem. Rev. 1988, 88, 541–562. [Google Scholar] [CrossRef]
- Miller, R.D.; Michl, J. Polysilane high polymers. Chem. Rev. 1989, 89, 1359–1410. [Google Scholar] [CrossRef]
- Jaeger, R.D.; Gleria, M. Poly(organophosphazene)s and related compounds: Synthesis, properties and applications. Prog. Polym. Sci. 1998, 23, 179–276. [Google Scholar] [CrossRef]
- He, X.; Baumgartner, T. Conjugated main-group polymers for optoelectronics. RSC Adv. 2013, 3, 11334–11350. [Google Scholar] [CrossRef]
- Wilfert, S.; Henke, H.; Schoefberger, W.; Brüggemann, O.; Teasdale, I. Chain-End-Functionalized Polyphosphazenes via a One-Pot-Phosphine Mediated Living Polymerization. Macromol. Rapid Commun. 2014, 35, 1135–1141. [Google Scholar] [CrossRef]
- Rawe, B.W.; Chun, C.P.; Gates, D.P. Anionic polymerisation of phosphaalkenes bearing polyaromatic chromophores: Phosphine polymers showing “turn-on” emission selectively with peroxide. Chem. Sci. 2014, 5, 4928–4938. [Google Scholar] [CrossRef]
- Fazen, P.J.; Beck, J.S.; Lynch, A.T.; Remsen, E.E.; Sneddon, L.G. Thermally induced borazine dehydropolymerization reactions. Synthesis and ceramic conversion reactions of a new high-yield polymeric precursor to boron nitride. Chem. Mater. 1990, 2, 96–97. [Google Scholar] [CrossRef]
- Jäkle, F. Advances in the synthesis of organoborane polymers for optical, electronic, and sensory applications. Chem. Rev. 2010, 110, 3985–4022. [Google Scholar] [CrossRef]
- Kuhtz, H.; Cheng, F.; Schwedler, S.; Bo, L.; Brockhinke, A.; Weber, L.; Parab, K.; Ja, F. Luminescent diazaborolyl-functionalized polystyrene. ACS Macro Lett. 2012, 1, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Hudson, Z.M.; Lunn, D.J.; Winnik, M.A.; Manners, I. Colour-tunable fluorescent multiblock micelles. Nat. Commun. 2014, 5, 3372. [Google Scholar] [CrossRef] [PubMed]
- Lorbach, A.; Bolte, M.; Li, H.; Lerner, H.W.; Holthausen, M.C.; Jäkle, F.; Wagner, M. 9, 10-Dihydro-9, 10-diboraanthracene: Supramolecular Structure and Use as a Building Block for Luminescent Conjugated Polymers. Angew. Chem. Int. Ed. 2009, 48, 4584–4588. [Google Scholar] [CrossRef] [PubMed]
- Allcock, H.R.; Chen, C. Polyphosphazenes: Phosphorus in inorganic–organic polymers. J. Org. Chem. 2020, 85, 14286–14297. [Google Scholar] [CrossRef]
- Vidal, F.; Jäkle, F. Functional polymeric materials based on main-group elements. Angew. Chem. Int. Ed. 2019, 58, 5846–5870. [Google Scholar] [CrossRef] [PubMed]
- Hübner, A.; Qu, Z.-W.; Englert, U.; Bolte, M.; Lerner, H.-W.; Holthausen, M.C.; Wagner, M. Main-chain boron-containing oligophenylenes via ring-opening polymerization of 9-H-9-borafluorene. J. Am. Chem. Soc. 2011, 133, 4596–4609. [Google Scholar] [CrossRef] [PubMed]
- Imori, T.; Lu, V.; Cai, H.; Tilley, T.D. Metal-catalyzed dehydropolymerization of secondary stannanes to high molecular weight polystannanes. J. Am. Chem. Soc. 1995, 117, 9931–9940. [Google Scholar] [CrossRef]
- Staubitz, A.; Presa, A.; Manners, I. Iridium-catalyzed dehydrocoupling of primary amine–borane adducts: A route to high molecular weight polyaminoboranes, boron–nitrogen analogues of polyolefins. Angew. Chem. Int. Ed. 2008, 47, 6212–6215. [Google Scholar] [CrossRef]
- Vance, J.R.; Robertson, A.P.; Lee, K.; Manners, I. Photoactivated, Iron-Catalyzed Dehydrocoupling of Amine–Borane Adducts: Formation of Boron–Nitrogen Oligomers and Polymers. Chem. Eur. J. 2011, 17, 4099–4103. [Google Scholar] [CrossRef] [PubMed]
- Staubitz, A.; Sloan, M.E.; Robertson, A.P.M.; Friedrich, A.; Schneider, S.; Gates, P.J.; der Günne, J.S.A.; Manners, I. Catalytic Dehydrocoupling/Dehydrogenation of N-Methylamine-Borane and Ammonia-Borane: Synthesis and Characterization of High Molecular Weight Polyaminoboranes. J. Am. Chem. Soc. 2010, 132, 13332–13345. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, B.L.; Goldberg, K.I.; Heinekey, D.M.; Autrey, T.; Linehan, J.C. Iridium-catalyzed dehydrogenation of substituted amine boranes: Kinetics, thermodynamics, and implications for hydrogen storage. Inorg. Chem. 2008, 47, 8583–8585. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.C.; Robertson, A.P.; Chaplin, A.B.; Sewell, L.J.; Thompson, A.L.; Haddow, M.F.; Manners, I.; Weller, A.S. Catching the First Oligomerization Event in the Catalytic Formation of Polyaminoboranes: H3B·NMeHBH2·NMeH2 Bound to Iridium. J. Am. Chem. Soc. 2011, 133, 11076–11079. [Google Scholar] [CrossRef]
- Johnson, H.C.; Weller, A.S. P-C-Activated Bimetallic Rhodium Xantphos Complexes: Formation and Catalytic Dehydrocoupling of Amine–Boranes. Angew. Chem. Int. Ed. 2015, 54, 10173–10177. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.C.; Leitao, E.M.; Whittell, G.R.; Manners, I.; Lloyd-Jones, G.C.; Weller, A.S. Mechanistic Studies of the Dehydrocoupling and Dehydropolymerization of Amine–Boranes Using a [Rh(Xantphos)]+ Catalyst. J. Am. Chem. Soc. 2014, 136, 9078–9093. [Google Scholar] [CrossRef]
- Colebatch, A.L.; Weller, A.S. Amine–Borane dehydropolymerization: Challenges and opportunities. Chem. Eur. J. 2019, 25, 1379–1390. [Google Scholar] [CrossRef]
- Han, D.; Anke, F.; Trose, M.; Beweries, T. Recent advances in transition metal catalysed dehydropolymerisation of amine boranes and phosphine boranes. Coord. Chem. Rev. 2019, 380, 260–286. [Google Scholar] [CrossRef]
- Wirtz, L.; Lambert, J.; Morgenstern, B.; Schäfer, A. Cross-Dehydrocoupling of Amines and Silanes Catalyzed by Magnesocenophanes. Organometallics 2021, 40, 2108–2117. [Google Scholar] [CrossRef]
- Denis, J.-M.; Forintos, H.; Szelke, H.; Toupet, L.; Pham, T.-N.; Madec, P.-J.; Gaumont, A.-C. B(C6F5)3-catalyzed formation of B–P bonds by dehydrocoupling of phosphine–boranes. Chem. Commun. 2003, 54–55. [Google Scholar] [CrossRef] [PubMed]
- Dorn, H.; Singh, R.A.; Massey, J.A.; Nelson, J.M.; Jaska, C.A.; Lough, A.J.; Manners, I. Transition metal-catalyzed formation of phosphorus− boron bonds: A new route to phosphinoborane rings, chains, and macromolecules. J. Am. Chem. Soc. 2000, 122, 6669–6678. [Google Scholar] [CrossRef]
- Dorn, H.; Singh, R.A.; Massey, J.A.; Lough, A.J.; Manners, I. Rhodium-Catalyzed Formation of Phosphorus–Boron Bonds: Synthesis of the First High Molecular Weight Poly (phosphinoborane). Angew. Chem. Int. Ed. 1999, 38, 3321–3323. [Google Scholar] [CrossRef]
- Dorn, H.; Rodezno, J.M.; Brunnhöfer, B.; Rivard, E.; Massey, J.A.; Manners, I. Synthesis, Characterization, and Properties of the Polyphosphinoboranes [RPH−BH2]n (R = Ph, iBu, p-nBuC6H4, p-dodecylC6H4): Inorganic Polymers with a Phosphorus−Boron Backbone. Macromolecules 2003, 36, 291–297. [Google Scholar] [CrossRef]
- Clark, T.J.; Rodezno, J.M.; Clendenning, S.B.; Aouba, S.; Brodersen, P.M.; Lough, A.J.; Ruda, H.E.; Manners, I. Rhodium-Catalyzed Dehydrocoupling of Fluorinated Phosphine–Borane Adducts: Synthesis, Characterization, and Properties of Cyclic and Polymeric Phosphinoboranes with Electron-Withdrawing Substituents at Phosphorus. Chem. Eur. J. 2005, 11, 4526–4534. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Lönnecke, P.; Hey-Hawkins, E. Phosphorus–Boron-Based Polymers Obtained by Dehydrocoupling of Ferrocenylphosphine–Borane Adducts. Eur. J. Inorg. Chem. 2014, 2014, 2456–2465. [Google Scholar] [CrossRef]
- Jacquemin, D.; Lambert, C.; Perpète, E.A. Structures and properties of polyphosphinoborane: An oligomeric theoretical study. Macromolecules 2004, 37, 1009–1015. [Google Scholar] [CrossRef]
- Schäfer, A.; Jurca, T.; Turner, J.; Vance, J.R.; Lee, K.; Du, V.A.; Haddow, M.F.; Whittell, G.R.; Manners, I. Iron-Catalyzed Dehydropolymerization: A Convenient Route to Poly (phosphinoboranes) with Molecular-Weight Control. Angew. Chem. Int. Ed. 2015, 54, 4836–4841. [Google Scholar] [CrossRef] [PubMed]
- Cavaye, H.; Clegg, F.; Gould, P.J.; Ladyman, M.K.; Temple, T.; Dossi, E. Primary alkylphosphine–borane polymers: Synthesis, low glass transition temperature, and a predictive capability thereof. Macromolecules 2017, 50, 9239–9248. [Google Scholar] [CrossRef]
- Marquardt, C.; Jurca, T.; Schwan, K.-C.; Stauber, A.; Virovets, A.V.; Whittell, G.R.; Manners, I.; Scheer, M. Metal-Free Addition/Head-to-Tail Polymerization of Transient Phosphinoboranes, RPH-BH2: A Route to Poly(alkylphosphinoboranes). Angew. Chem. Int. Ed. 2015, 54, 13782–13786. [Google Scholar] [CrossRef]
- Stauber, A.; Jurca, T.; Marquardt, C.; Fleischmann, M.; Seidl, M.; Whittell, G.R.; Manners, I.; Scheer, M. A Convenient Route to Monoalkyl-Substituted Phosphanylboranes (HRP–BH2–NMe3): Prospective Precursors to Poly [(alkylphosphino) boranes]. Eur. J. Inorg. Chem. 2016, 2016, 2684–2687. [Google Scholar] [CrossRef]
- Lehnfeld, F.; Seidl, M.; Timoshkin, A.Y.; Scheer, M. Synthesis and Reactivity of a Lewis-Base-Stabilized tert-Butyl Arsanylborane: A Versatile Building Block for Arsenic-Boron Oligomers. Eur. J. Inorg. Chem. 2022, 2022, e202100930. [Google Scholar] [CrossRef]
- Braese, J. Reactivity of Lewis Base Stabilized Pnictogenylboranes Towards Transition Metal Complexes. Ph.D. Thesis, University of Regensburg, Regensburg, Germany, 2019. [Google Scholar]
- Braese, J.; Lehnfeld, F.; Annibale, V.; Oswald, T.; Beckhaus, R.; Manners, I.; Scheer, M. Titanium-Catalyzed Polymerization of a Lewis Base-Stabilized Phosphinoborane. Chem. Eur. J. 2023, 29, e202301741. [Google Scholar] [CrossRef]
- Vogel, U.; Hoemensch, P.; Schwan, K.-C.; Timoshkin, A.Y.; Scheer, M. The Stabilisation of Monomeric Parent Compounds of Phosphanyl-and Arsanylboranes. Chem. Eur. J. 2003, 9, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Langhans, K.P.; Stelzer, O.; Svara, J.; Weferling, N. Synthesis of Primary and Secondary Phosphines by Selective Alkylation of PH3 under Phase Transfer Condition. Z. Naturforsch. B 1990, 45, 203–211. [Google Scholar] [CrossRef]
- Diekmann, M.; Bockstiegel, G.; Lützen, A.; Friedemann, M.; Saak, W.; Haase, D.; Beckhaus, R. Chiral Bis(η5:η1-pentafulvene)titanium Complexes. Organometallics 2006, 25, 339–348. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Substituent | δ (31P) [a] | δ (11B) [a] | Yield [b] |
|---|---|---|---|---|
| 1a | n-propyl | −130.4 | −3.2 | 28%/29% |
| 1b | n-butyl | −127.6 | −4.0 | 13%/54% |
| 1c | n-hexyl | −128.7 | −3.3 | 32%/66% |
| Catalyst Loading | Reaction Time | Temperature | Concentration 1a [mol/L] | Conversion |
|---|---|---|---|---|
| - | 90 min | r.t. | 0.089 | 19% |
| - | 24 h | r.t. | 0.089 | 23% |
| 5 mol% | 90 min | r.t. | 0.089 | 41% |
| 5 mol% | 24 h | r.t. | 0.089 | 51% |
| 10 mol% | 24 h | r.t. | 0.089 | 64% |
| 10 mol% | 210 min | r.t. | 0.03 | 54% |
| 10 mol% | 42 h | r.t.. | 0.03 | 76% |
| Catalyst Loading | Reaction Time | Temperature | Concentration 1c [mol/L] | Conversion |
|---|---|---|---|---|
| - | 16 h | r.t. | 0.1 | 18% |
| - | 16 h | 323 K | Neat [a] | 84% |
| - | 40 h | 323 K | Neat [a] | 97% |
| 10 mol% | 3 h | r.t. | 0.1 | 17% |
| 10 mol% | 3 h | r.t. | 0.2 | 27% |
| 4 mol% | 30 min | r.t. | 0.4 | 18% |
| 4 mol% | 90 min | r.t. | 0.4 | 22% |
| 4 mol% | 7 d | r.t. | 0.4 | 41% |
| 4 mol% | 21 d | r.t. | 0.4 | 65% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehnfeld, F.; Oswald, T.; Beckhaus, R.; Scheer, M. Mono-Alkyl-Substituted Phosphinoboranes (HRP–BH2–NMe3) as Precursors for Poly(alkylphosphinoborane)s: Improved Synthesis and Comparative Study. Inorganics 2023, 11, 377. https://doi.org/10.3390/inorganics11100377
Lehnfeld F, Oswald T, Beckhaus R, Scheer M. Mono-Alkyl-Substituted Phosphinoboranes (HRP–BH2–NMe3) as Precursors for Poly(alkylphosphinoborane)s: Improved Synthesis and Comparative Study. Inorganics. 2023; 11(10):377. https://doi.org/10.3390/inorganics11100377
Chicago/Turabian StyleLehnfeld, Felix, Tim Oswald, Rüdiger Beckhaus, and Manfred Scheer. 2023. "Mono-Alkyl-Substituted Phosphinoboranes (HRP–BH2–NMe3) as Precursors for Poly(alkylphosphinoborane)s: Improved Synthesis and Comparative Study" Inorganics 11, no. 10: 377. https://doi.org/10.3390/inorganics11100377
APA StyleLehnfeld, F., Oswald, T., Beckhaus, R., & Scheer, M. (2023). Mono-Alkyl-Substituted Phosphinoboranes (HRP–BH2–NMe3) as Precursors for Poly(alkylphosphinoborane)s: Improved Synthesis and Comparative Study. Inorganics, 11(10), 377. https://doi.org/10.3390/inorganics11100377