
A Single Biaryl Monophosphine Ligand Motif—The Multiverse of Coordination Modes





Abstract
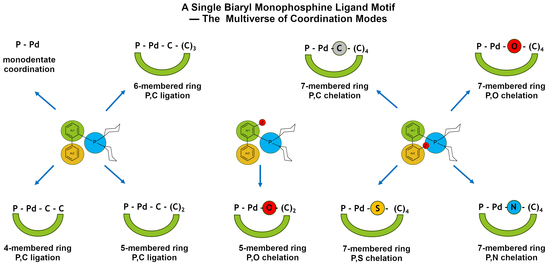
:
1. Introduction
2. Results
2.1. Analysis of Crystalographic Data Recorded for Single Crystals of 9–12
2.1.1. C,P Chelation in Five-Membered Palladacycle 9
2.1.2. O,P Chelation with Simultaneous Demethylation to Form Five-Membered Palladacycle 10
2.1.3. Additional Monodentate P Ligation by Five-Membered Palladacycle 11
2.1.4. Monodentate S ligation of Biaryl Monophosphine Sulfide 12
2.1.5. Monodentate P Ligation of Nap-Phos in 14 with Unusual C–H…Pd Contacts
2.2. Multiverse of Coordination Modes of Biaryl Monophosphine Ligands Based on the CSD Survey
2.2.1. C,P Chelation—Four- and Five-Membered Metallacycles
Four-Membered Palladacycles
Five-Membered Palladacycles
Six-Membered Palladacycles
2.2.2. Chelation via Heteroatoms Forming Seven-Membered Rings
2.2.3. Monodentate P Ligation
2.3. Coordination Modef of Phosphine Sulfides
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of Dicyclohexyl[1,4-dimethoxy-3-(2,4,6-trimethoxyphenyl)naphthalen-2-yl]phosphane sulfide (5)
3.1.2. Synthesis of Dicyclohexyl[1,4-dimethoxy-3-(2,4,6-trimethoxyphenyl)naphthalen-2-yl]phosphane (1)
3.1.3. Synthesis of Chloro(dicyclohexyl)(1,4-dimethoxy-3-phenylnaphthalen-2-yl)phosphonium chloride (7)
3.1.4. Transformation of 7 to 5
3.1.5. Transformation of 7 to Dicyclohexyl[1,4-dimethoxy-3-(2,4,6-trimethoxyphenyl)naphthalen-2-yl]phosphane borane 8
3.1.6. Transformation of 7 to Dicyclohexyl[1,4-dimethoxy-3-(2,4,6-trimethoxyphenyl)naphthalen-2-yl]phosphane 1
3.1.7. Transformation of 8 to 1
3.1.8. Synthesis of Complex 9
3.1.9. Synthesis of Complex 10
3.1.10. Synthesis of Complex 11
3.1.11. Synthesis of Complex 12
3.1.12. Synthesis of Complex 14
3.2. Crystallography
4. Conclusions
- (i)
- Under mild conditions (35 °C), a C,P chelated Pd(II) complex of 9 is formed;
- (ii)
- At a slightly higher temperature (40 °C), a by-product complex 10 is obtained through demethylation of one of the methoxy groups at the 1,4-dimethoxynaphthalene fragment;
- (iii)
- 11 can serve as an example of an intermediate stage of P-methylation;
- (iv)
- 12 is the first report on a palladium(II) coordination compound of biaryl monophosphine sulfide and shows monodentate S ligation.
Author Contributions
Funding
Conflicts of Interest
References
- Dotta, P.; Kumar, P.G.A.; Pregosin, P.S.; Albinati, A.; Rizzato, S. Pd-(MOP) chemistry: Novel bonding modes and interesting charge distribution. Organometallics 2003, 22, 5345–5349. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Kaplon, K.; Mazur, L.; Strzelecka, D.; Pietrusiewicz, K.M. Readily available catalysts for demanding Suzuki-Miyaura couplings under mild conditions. Tetrahedron 2016, 72, 6668–6677. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Kielar, K.; Pietrusiewicz, K.M. Rational design of novel ligands for environmentally benign cross-coupling reactions. Pure Appl. Chem. 2011, 83, 633–644. [Google Scholar] [CrossRef]
- Barder, T.E.; Walker, S.D.; Martinelli, J.R.; Buchwald, S.L. Catalysts for Suzuki-Miyaura coupling processes: Scope and studies of the effect of ligand structure. J. Am. Chem. Soc. 2005, 127, 4685–4696. [Google Scholar] [CrossRef]
- Altman, R.A.; Buchwald, S.L. Pd-catalyzed Suzuki-Miyaura reactions of aryl halides using bulky biarylmonophosphine ligands. Nat. Protoc. 2007, 2, 3115–3121. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, K.; Frynas, S.; Miroslaw, B.; Lipkowski, J.; Demchuk, O.M. An Efficient Asymmetric Cross-Coupling Reaction in Aqueous Media Mediated by Chiral Chelating Mono Phosphane Atropisomeric Biaryl Ligand. Catalysts 2023, 13, 353. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Arlt, D.; Jasinski, R.; Pietrusiewicz, K.M. Relationship between structure and efficiency of atropisomeric phosphine ligands in homogeneous catalytic asymmetric hydrogenation. J. Phys. Org. Chem. 2012, 25, 1006–1011. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Martyna, A.; Kwasnik, M.; Szwaczko, K.; Strzelecka, D.; Miroslaw, B.; Pietrusiewicz, K.M.; Urbanczyk-Lipkowska, Z. A Modular Approach to Atropisomeric Bisphosphines of Diversified Electronic Density on Phosphorus Atoms. Molecules 2022, 27, 5504. [Google Scholar] [CrossRef]
- Jasinski, R.; Demchuk, O.M.; Babyuk, D. A Quantum-Chemical DFT Approach to Elucidation of the Chirality Transfer Mechanism of the Enantioselective Suzuki-Miyaura Cross-Coupling Reaction. J. Chem. 2017, 2017, 3617527. [Google Scholar] [CrossRef]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C-N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Singh, C.; Rathod, J.; Jha, V.; Panossian, A.; Kumar, P.; Leroux, F.R. Modular Synthesis of Biaryl-Substituted Phosphine Ligands: Application in Microwave-Assisted Palladium-Catalyzed C-N Cross-Coupling Reactions. Eur. J. Org. Chem. 2015, 2015, 6515–6525. [Google Scholar] [CrossRef]
- Xu, J.; Liu, R.Y.; Yeung, C.S.; Buchwald, S.L. Monophosphine Ligands Promote Pd-Catalyzed C-S Cross-Coupling Reactions at Room Temperature with Soluble Bases. ACS Catal. 2019, 9, 6461–6466. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Zahoor, A.F.; Ahmad, S.; Noreen, R.; Khan, S.G.; Ahmad, H. Cross-coupling reactions towards the synthesis of natural products. Mol. Divers. 2022, 26, 647–689. [Google Scholar] [CrossRef] [PubMed]
- Buskes, M.J.; Blanco, M.J. Impact of Cross-Coupling Reactions in Drug Discovery and Development. Molecules 2020, 25, 3493. [Google Scholar] [CrossRef]
- Devendar, P.; Qu, R.Y.; Kang, W.M.; He, B.; Yang, G.F. Palladium-Catalyzed Cross-Coupling Reactions: A Powerful Tool for the Synthesis of Agrochemicals. J. Agric. Food Chem. 2018, 66, 8914–8934. [Google Scholar] [CrossRef]
- Horbaczewskyj, C.S.; Fairlamb, I.J.S. Pd-Catalyzed Cross-Couplings: On the Importance of the Catalyst Quantity Descriptors, mol % and ppm. Org. Process. Res. Dev. 2022, 26, 2240–2269. [Google Scholar] [CrossRef]
- McCarthy, S.; Braddock, D.C.; Wilton-Ely, J.D.E.T. Strategies for sustainable palladium catalysis. Coord. Chem. Rev. 2021, 442, 213925. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.X.; Liu, C.B.; Zhang, B.; Du, Y.H.; Liu, C.F.; Ma, L.; Xi, S.B.; Li, R.L.; Zhao, X.X.; et al. Molecular engineered palladium single atom catalysts with an M-C1N3 subunit for Suzuki coupling. J. Mater. Chem. A 2021, 9, 11427–11432. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Yoruk, B.; Blackburn, T.; Snieckus, V. A mixed naphthyl-phenyl phosphine ligand motif for Suzuki, Heck, and hydrodehalogenation reactions. Synlett 2006, 2006, 2908–2913. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Demchuk, O.M.; Swierczynska, W.; Dziuba, K.; Frynas, S.; Flis, A.; Pietrusiewicz, K.M. Raney-Ni reduction of phosphine sulfides. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 64–68. [Google Scholar] [CrossRef]
- Dybala, I.; Demchuk, O.M. Tris(acetonitrile)chloropalladium tetrafluoroborate synthesis, application and structural analysis. J. Mol. Struct. 2016, 1121, 135–141. [Google Scholar] [CrossRef]
- Sajjad, M.A.; Christensen, K.E.; Rees, N.H.; Schwerdtfeger, P.; Harrison, J.A.; Nielson, A.J. New complexity for aromatic ring agostic interactions. Chem. Commun. 2017, 53, 4187–4190. [Google Scholar] [CrossRef]
- Lin, X.; Wu, W.; Mo, Y. Agostic Interactions in Early Transition-Metal Complexes: Roles of Hyperconjugation, Dispersion, and Steric Effect. Chem. Eur. J. 2019, 25, 6591. [Google Scholar] [CrossRef]
- Brookhart, M.; Green, M.L.H.; Parkin, G. Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. USA 2007, 104, 6908–6914. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Biradha, K.; Desiraju, G.R. Agostic interactions in organometallic compounds. A Cambridge Structural Database study. J. Chem. Soc. Dalton Trans. 1996, 20, 3925–3930. [Google Scholar] [CrossRef]
- Hupf, E.; Malaspina, L.A.; Holsten, S.; Kleemiss, F.; Edwards, A.J.; Price, J.R.; Kozich, V.; Heyne, K.; Mebs, S.; Grabowsky, S.; et al. Proximity Enforced Agostic Interactions Involving Closed-Shell Coinage Metal Ions. Inorg. Chem. 2019, 58, 16372–16378. [Google Scholar] [CrossRef]
- Grubbs, R.H.; Coates, G.W. α-Agostic Interactions and Olefin Insertion in Metallocene Polymerization Catalysts. Acc. Chem. Res. 1996, 29, 85–93. [Google Scholar] [CrossRef]
- Xu, C.; Li, G.; Etienne, M.; Leng, X.; Chen, Y. α-C–C agostic interactions and C–H bond activation in scandium cyclopropyl complexes. Inorg. Chem. Front. 2020, 7, 4822–4831. [Google Scholar] [CrossRef]
- Moss, G.P.; Smith, P.A.S.; Tavernier, D. Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995). Pure Appl. Chem. 1995, 67, 1307–1375. [Google Scholar] [CrossRef]
- Salzer, A. Nomenclature of Organometallic Compounds of the Transition Elements. Pure Appl. Chem. 1999, 71, 1557–1585. [Google Scholar] [CrossRef]
- Cook, T.R.; Stang, P.J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Du, J.; Olenyuk, B.; Stang, P.J.; Sun, Y. The Applications of Metallacycles and Metallacages. Inorganics 2023, 11, 54. [Google Scholar] [CrossRef]
- Sun, Y.; Stang, P.J. Metallacycles, metallacages, and their aggregate/optical behavior. Aggregate 2021, 2, e94. [Google Scholar] [CrossRef]
- Bosque, R.; Maseras, F. A theoretical assessment of the thermodynamic preferences in the cyclopalladation of amines. Eur. J. Inorg. Chem. 2005, 2005, 4040–4047. [Google Scholar] [CrossRef]
- Montgomery, M.; O’Brien, H.M.; Mendez-Galvez, C.; Bromfield, C.R.; Roberts, J.P.M.; Winnicka, A.M.; Horner, A.; Elorriaga, D.; Sparkes, H.A.; Bedford, R.B. The surprisingly facile formation of Pd(i)-phosphido complexes from ortho-biphenylphosphines and palladium acetate. Dalton Trans. 2019, 48, 3539–3542. [Google Scholar] [CrossRef]
- Allgeier, A.M.; Shaw, B.J.; Hwang, T.L.; Milne, J.E.; Tedrow, J.S.; Wilde, C.N. Characterization of Two Stable Degradants of Palladium (t)BuXPhos Catalyst and a Unique Dearomatization Reaction. Organometallics 2012, 31, 519–522. [Google Scholar] [CrossRef]
- Christmann, U.; Pantazis, D.A.; Benet-Buchholz, J.; McGrady, J.E.; Maseras, F.; Vilar, R. Experimental and theoretical investigations of new dinuclear palladium complexes as precatalysts for the amination of aryl chlorides. J. Am. Chem. Soc. 2006, 128, 6376–6390. [Google Scholar] [CrossRef] [PubMed]
- Toriumi, N.; Shimomaki, K.; Caner, J.; Murata, K.; Martin, R.; Iwasawa, N. Mechanistic Studies into Visible Light-Driven Carboxylation of Aryl Halides/Triflates by the Combined Use of Palladium and Photoredox Catalysts. Bull. Chem. Soc. Jpn. 2021, 94, 1846–1853. [Google Scholar] [CrossRef]
- Lalloo, N.; Malapit, C.A.; Taimoory, S.M.; Brigham, C.E.; Sanford, M.S. Decarbonylative Fluoroalkylation at Palladium(II): From Fundamental Organometallic Studies to Catalysis. J. Am. Chem. Soc. 2021, 143, 18617–18625. [Google Scholar] [CrossRef]
- Pratap, R.; Parrish, D.; Gunda, P.; Venkataraman, D.; Lakshman, M.K. Influence of Biaryl Phosphine Structure on C-N and C-C Bond Formation. J. Am. Chem. Soc. 2009, 131, 12240–12249. [Google Scholar] [CrossRef]
- Shen, X.Q.; Jones, G.O.; Watson, D.A.; Bhayana, B.; Buchwald, S.L. Enantioselective Synthesis of Axially Chiral Biaryls by the Pd-Catalyzed Suzuki Miyaura Reaction: Substrate Scope and Quantum Mechanical Investigations. J. Am. Chem. Soc. 2010, 132, 11278–11287. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Boursalian, G.B.; Ritter, T. Palladium-Catalyzed Decarbonylative Difluoromethylation of Acid Chlorides at Room Temperature. Angew. Chem.-Int. Ed. 2018, 57, 16871–16876. [Google Scholar] [CrossRef] [PubMed]
- Bruno, N.C.; Tudge, M.T.; Buchwald, S.L. Design and preparation of new palladium precatalysts for C-C and C-N cross-coupling reactions. Chem. Sci. 2013, 4, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Sivendran, N.; Pirkl, N.; Hu, Z.Y.; Doppiu, A.; Goossen, L.J. Halogen-Bridged Methylnaphthyl Palladium Dimers as Versatile Catalyst Precursors in Coupling Reactions. Angew. Chem.-Int. Ed. 2021, 60, 25151–25160. [Google Scholar] [CrossRef] [PubMed]
- Biscoe, M.R.; Barder, T.E.; Buchwald, S.L. Electronic effects on the selectivity of Pd-catalyzed C-N bond-forming reactions using biarylphosphine ligands: The competitive roles of amine binding and acidity. Angew. Chem.-Int. Ed. 2007, 46, 7232–7235. [Google Scholar] [CrossRef] [PubMed]
- Faller, J.W.; Wilt, J.C.; Parr, J. Kinetic resolution and unusual regioselectivity in palladium-catalyzed allylic alkylations with a chiral P,S ligand. Org. Lett. 2004, 6, 1301–1304. [Google Scholar] [CrossRef]
- Faller, J.W.; Wilt, J.C. Palladium/BINAP(S)-catalyzed asymmetric allylic amination. Org. Lett. 2005, 7, 633–636. [Google Scholar] [CrossRef]
- Gilbertson, S.R.; Wang, X.F. Synthesis of (dicyclohexylphosphino)serine, its incorporation into a dodecapeptide, and the coordination of rhodium. J. Org. Chem. 1996, 61, 434–435. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction Ltd. Crysalis-Pro Software System, Version 1.171.38.46; Rigaku Corporation Ltd.; Rigaku Oxford Diffraction Ltd.; Rigaku Corporation: Oxford, UK, 2016.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta. Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | 9 | 10 | 11 | 12 |
|---|---|---|---|---|
| Empirical formula | C71H98Cl6O11P2Pd2 | C66H86Cl2O10P2Pd | C65H87ClO12P2Pd | C87H110Cl4O10P2Pd2S2 |
| Formula weight | 1615.06 | 1278.68 | 1264.14 | 1796.41 |
| Temperature/K | 120 | 290 | 290 | 100 |
| Crystal system | monoclinic | monoclinic | triclinic | orthorhombic |
| Space group | C2/c | P21 | P-1 | Pbca |
| a/Å | 18.4535(2) | 11.6734(16) | 12.263(1) | 22.926(5) |
| b/Å | 15.1832(1) | 17.0922(19) | 13.884(1) | 12.246(2) |
| c/Å | 26.2221(2) | 16.7564(19) | 20.773(1) | 29.303(6) |
| α/° | 90 | 90 | 88.53(1) | 90 |
| β/° | 97.482(1) | 107.391(14) | 85.29(1) | 90 |
| γ/° | 90 | 90 | 65.98(1) | 90 |
| Volume/Å3 | 7284.44(11) | 3190.5(7) | 3219.5(5) | 8227(3) |
| Z | 4 | 2 | 2 | 4 |
| ρcalcg/cm3 | 1.4725 | 1.3309 | 1.304 | 1.450 |
| μ/mm−1 | 6.886 | 0.481 | 0.438 | 0.715 |
| F(000) | 3364.6 | 1343.6 | 1332.0 | 3736.0 |
| Crystal size/mm3 | 0.12 × 0.08 × 0.02 | 0.35 × 0.15 × 0.15 | 0.3 × 0.15 × 0.08 | 0.35 × 0.2 × 0.08 |
| Radiation | Cu Kα | Mo Kα | Mo Kα | Mo Kα |
| (λ = 1.54184) | (λ = 0.71073) | (λ = 0.71073) | (λ = 0.71073) | |
| 2Θ range for data collection/° | 6.8 to 135.36 | 5.4 to 50.48 | 5.16 to 50.48 | 4.68 to 50.48 |
| Index ranges | −22 ≤ h ≤ 22, −18 ≤ k ≤ 18, −28 ≤ l ≤ 31 | −13 ≤ h ≤ 14, −20 ≤ k ≤ 20, −20 ≤ l ≤ 20 | −14 ≤ h ≤ 14, −16 ≤ k ≤ 16, −24 ≤ l ≤ 24 | −16 ≤ h ≤ 27, −14 ≤ k ≤ 9, −35 ≤ l ≤ 32 |
| Reflections collected | 38,734 | 20,908 | 38,607 | 29,292 |
| Independent reflections | 6599 [Rint = 0.0408, Rsigma = 0.0261] | 7543 [Rint = 0.0728, Rsigma = 0.1356] | 11639 [Rint = 0.0963, Rsigma = 0.1492] | 7433 [Rint = 0.1199, Rsigma = 0.2016] |
| Data/restraints/parameters | 6599/0/428 | 7543/0/730 | 11,639/0/720 | 7433/7/427 |
| Goodness-of-fit on F2 | 1.05 | 0.971 | 1.302 | 0.825 |
| Final R indexes [I>=2σ (I)] | R1 = 0.0354 | R1 = 0.0595 | R1 = 0.0911 | R1 = 0.0513 |
| wR2 = 0.0792 | wR2 = 0.1138 | wR2 = 0.2315 | wR2 = 0.1236 | |
| Final R indexes [all data] | R1 = 0.0389 | R1 = 0.0595 | R1 = 0.1509 | R1 = 0.1307 |
| wR2 = 0.0819 | wR2 = 0.1138 | wR2 = 0.2502 | wR2 = 0.1691 | |
| Largest diff. peak/hole/e Å−3 | 1.48/−0.93 | 1.27/−0.65 | 1.64/−1.98 | 1.17/−1.22 |
| Flack parameter | – | −0.03(3) | – | – |
| CCDC No. | 2278033 | 2278034 | 2278032 | 2278031 |
| Complex | D-H…A | D-H | D...A | H...A | <D-H...A |
|---|---|---|---|---|---|
| 9 | C25-H25A…O4 i | 0.97 | 3.263(4) | 2.641(4) | 122(1) |
| C6-H6…Cl2 ii | 0.93 | 3.521(3) | 2.716(3) | 145(1) | |
| 10 | C38-H38…O9 iii | 0.93 | 3.46(1) | 2.67 | 143(1) |
| C43-H43A…O1 | 0.96 | 3.41(1) | 2.65 | 136(1) | |
| C47-H47…O2 iv | 0.93 | 3.55(1) | 2.63 | 170(1) | |
| 11 | C44-H44B…Pd1 | 0.96 | 3.36(1) | 2.73 | 124(1) |
| C62-H62B…Pd1 | 0.97 | 3.20(1) | 2.54 | 126(1) | |
| C21-H21B…O5 v | 0.96 | 3.456(18) | 2.65 | 142(1) | |
| C40-H40…O4 v | 0.93 | 3.375(16) | 2.56 | 147(1) | |
| 12 | C27-H27A…Pd1 | 1.00 | 3.322(7) | 2.42 | 152(2) |
| C21-H21A…O1 vi | 0.98 | 3.444(10) | 2.63 | 141(1) | |
| C8-H8…O5 vii | 0.95 | 3.492(9) | 2.62 | 153(1) | |
| C7-H7…O2 vii | 0.95 | 3.354(9) | 2.75 | 122(1) | |
| C40-H40B…O4 viii | 0.98 | 3.605(14) | 2.74 | 148(1) | |
| C40-H40A…Pd1 | 0.98 | 3.712(14) | 2.81 | 153(1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miroslaw, B.; Dybala, I.; Jasiński, R.; Demchuk, O.M. A Single Biaryl Monophosphine Ligand Motif—The Multiverse of Coordination Modes. Inorganics 2023, 11, 399. https://doi.org/10.3390/inorganics11100399
Miroslaw B, Dybala I, Jasiński R, Demchuk OM. A Single Biaryl Monophosphine Ligand Motif—The Multiverse of Coordination Modes. Inorganics. 2023; 11(10):399. https://doi.org/10.3390/inorganics11100399
Chicago/Turabian StyleMiroslaw, Barbara, Izabela Dybala, Radomir Jasiński, and Oleg M. Demchuk. 2023. "A Single Biaryl Monophosphine Ligand Motif—The Multiverse of Coordination Modes" Inorganics 11, no. 10: 399. https://doi.org/10.3390/inorganics11100399
APA StyleMiroslaw, B., Dybala, I., Jasiński, R., & Demchuk, O. M. (2023). A Single Biaryl Monophosphine Ligand Motif—The Multiverse of Coordination Modes. Inorganics, 11(10), 399. https://doi.org/10.3390/inorganics11100399