


Half-Sandwich Nickelacarboranes Derived from [7-(MeO(CH2)2S)-7,8-C2B9H11]−


 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
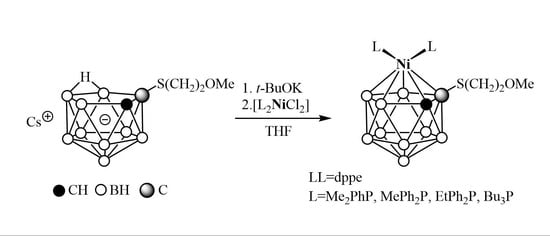
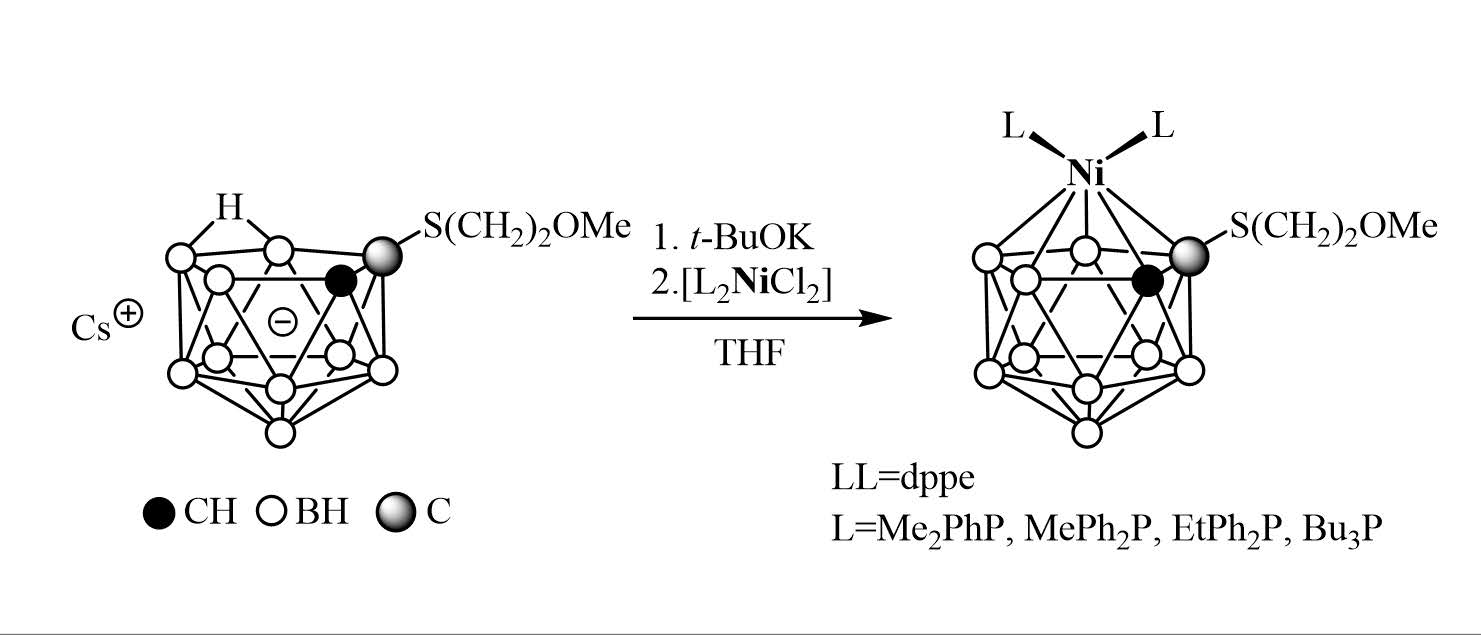
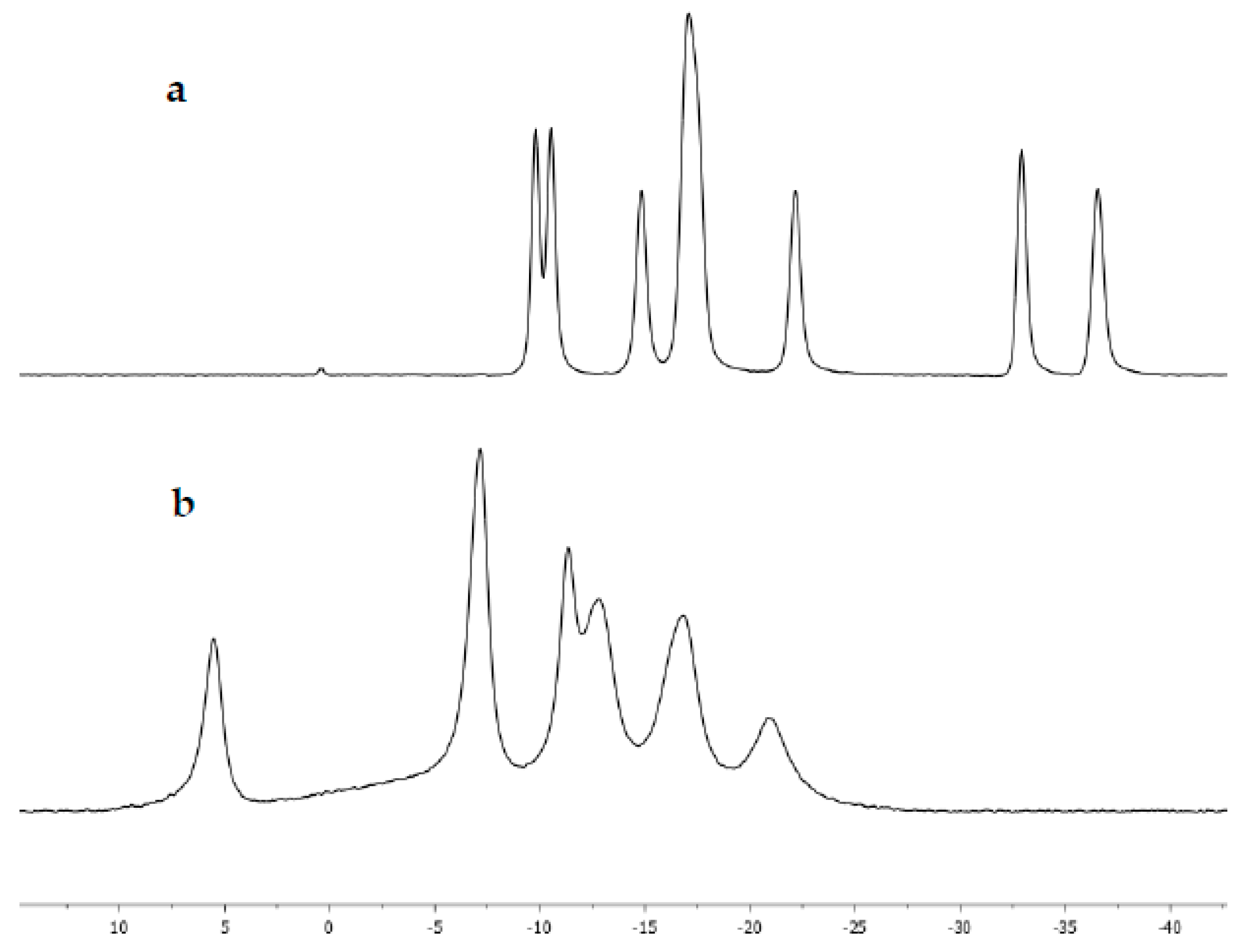
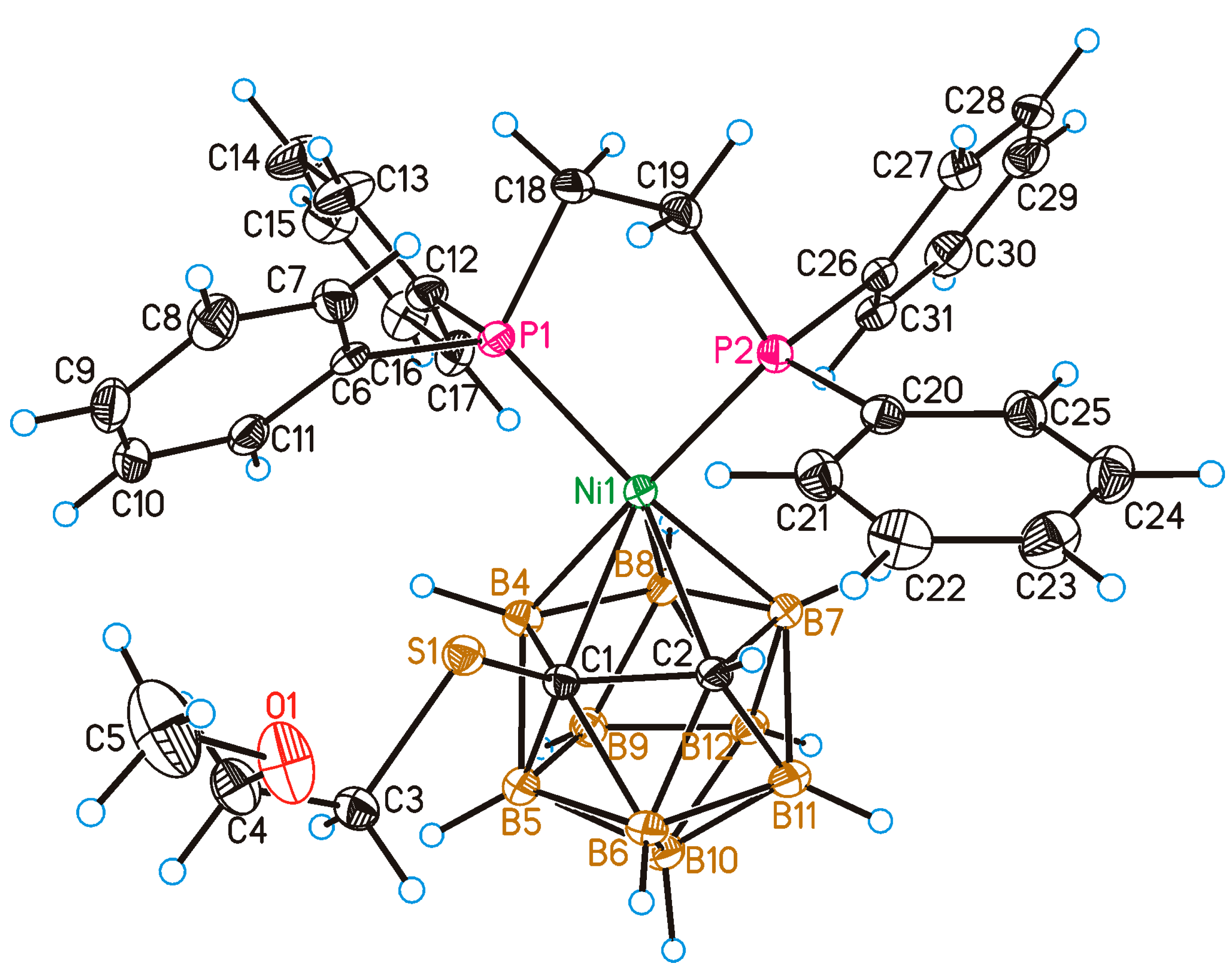
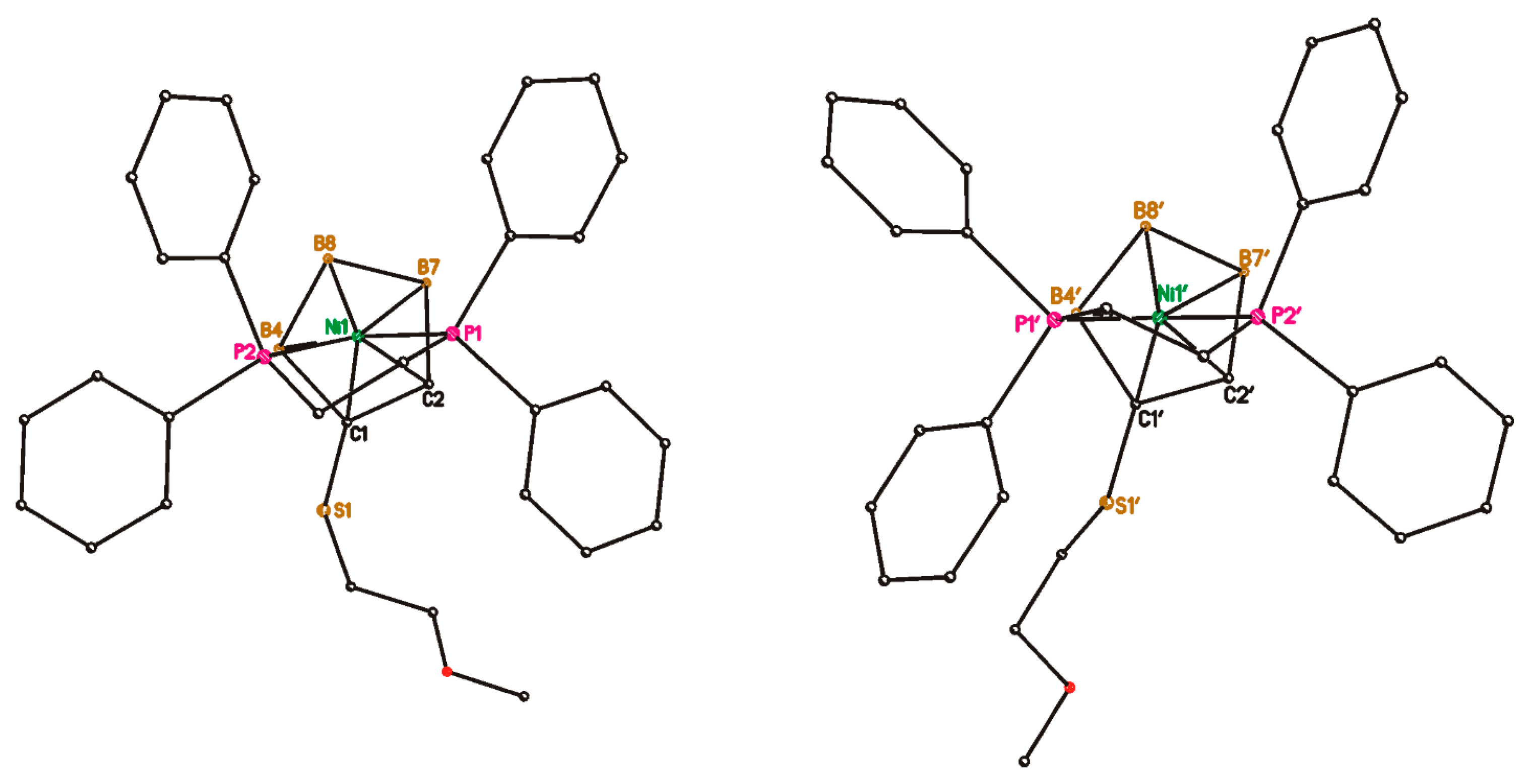
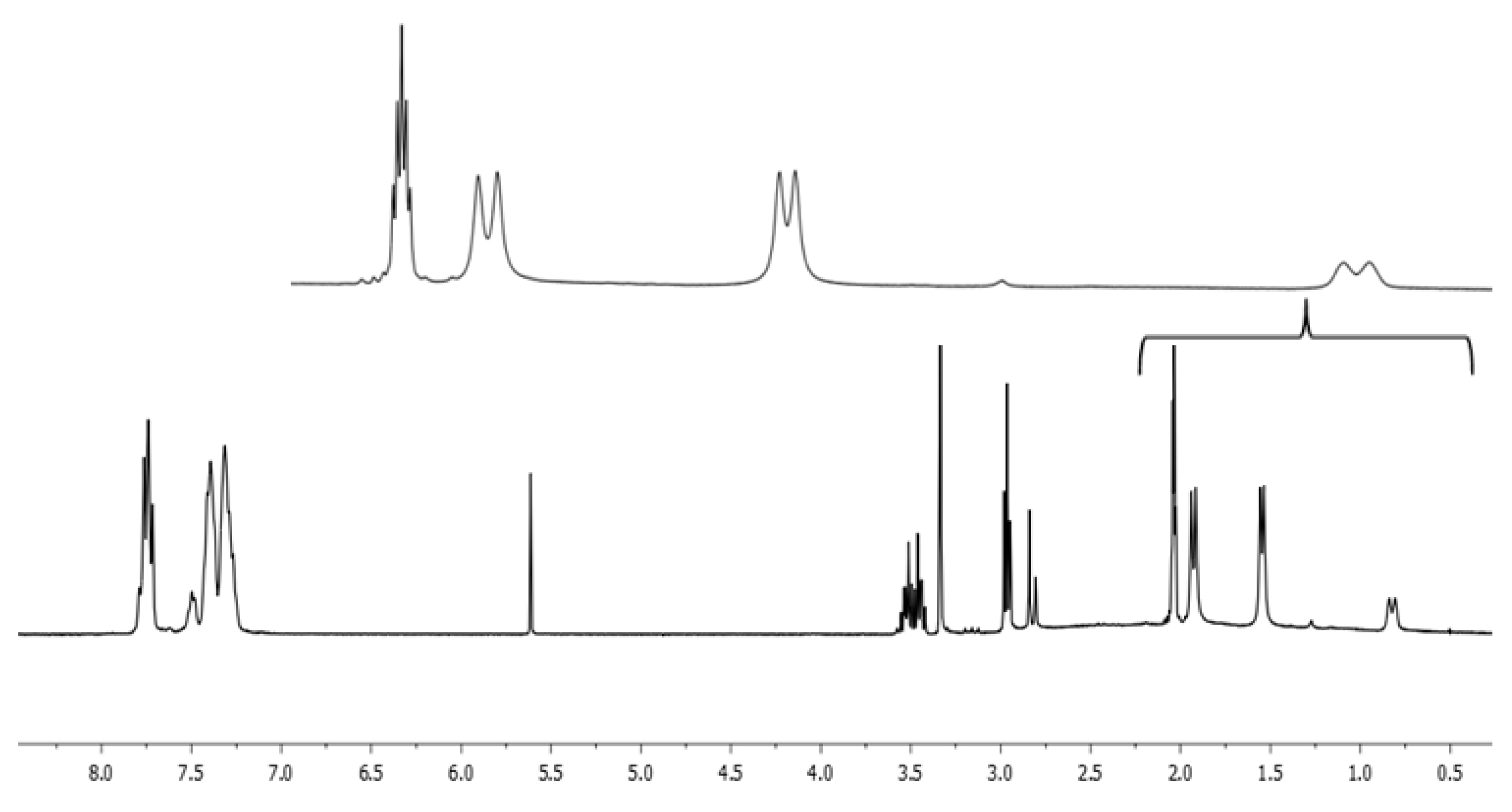
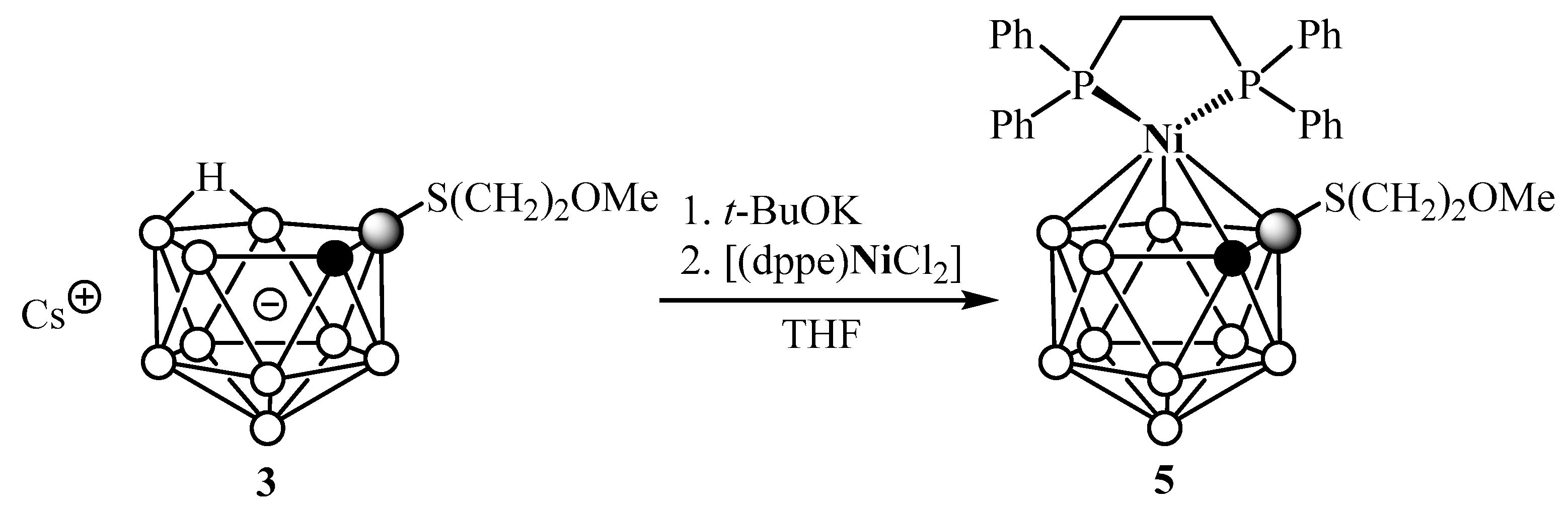
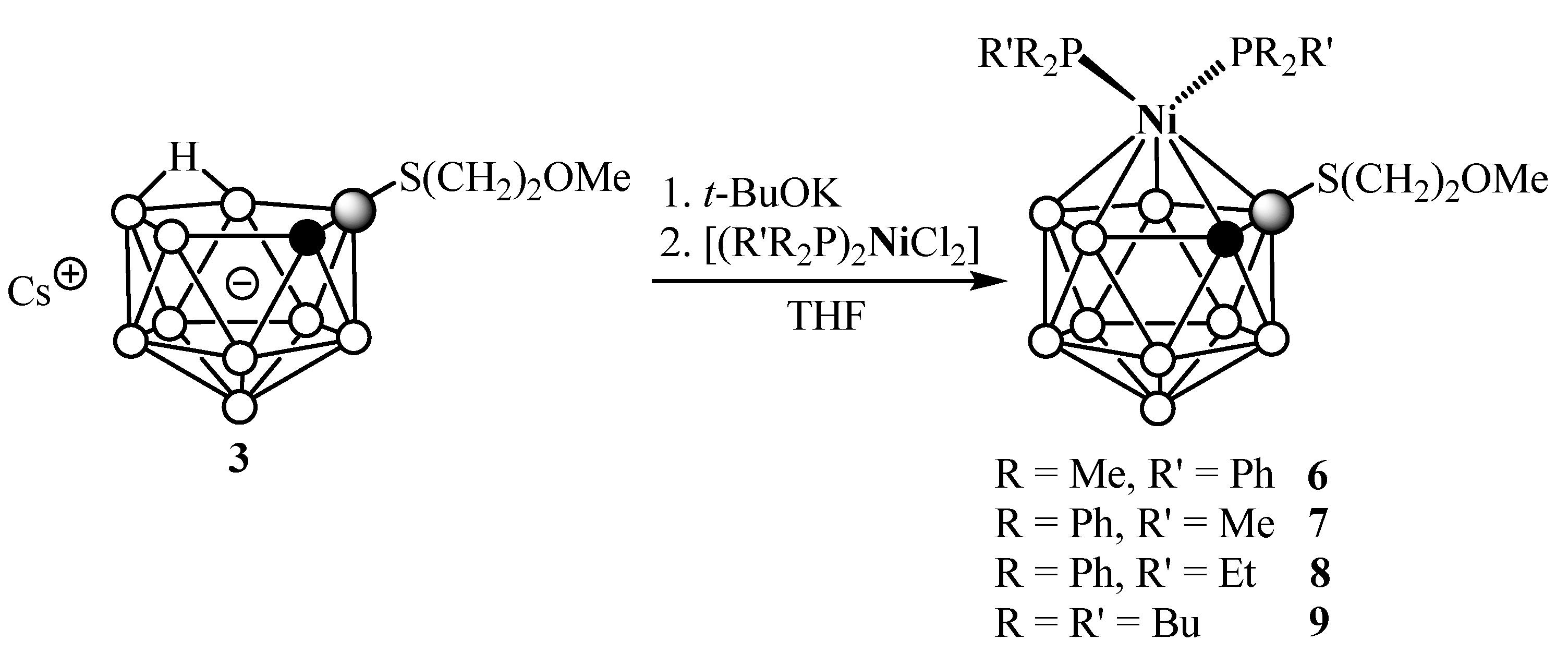
2. Results and Discussion
3. Conclusions
4. Experimental Section
4.1. Materials and Methods
4.2. Synthesis of 1-(MeO(CH2)2S)-1,2-C2B10H11 (1)
4.3. Synthesis of 1-(MeO(CH2)3S)-1,2-C2B10H11 (2)
4.4. Synthesis of Cs[7-(MeO(CH2)2S)-7,8-C2B9H11] (3)
4.5. Synthesis of Cs[7-(MeO(CH2)3S)-7,8-C2B9H11] (4)
4.6. General Procedure for the Synthesis of Complexes 5–9
4.7. Single Crystal X-ray Diffraction Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiesboeck, R.A.; Hawthorne, M.F. Dicarbaundecaborane(13) and derivatives. J. Am. Chem. Soc. 1964, 86, 1642–1643. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Young, D.C.; Garrett, P.M.; Owen, D.A.; Schwerin, S.G.; Tebbe, F.N.; Wegner, P.A. Preparation and characterization of the (3)-1,2- and (3)-1,7-dicarbadodecahydroundecaborate(-1) ions. J. Am. Chem. Soc. 1968, 90, 862–868. [Google Scholar] [CrossRef]
- Buchanan, J.; Hamilton, E.J.M.; Reed, D.; Welch, A.J. The structure of [7,8-C2B9H12]–; correction of a popular misconception. J. Chem. Soc. Dalton. Trans. 1990, 2, 677–680. [Google Scholar] [CrossRef]
- Harris, C.B. Transition metal nuclear quadrupole resonance. I. Cobalt-59 nuclear quadrupole resonance in Cs(1,2-B9C2H11)2Co(III) and the bonding in ferrocene. Inorg. Chem. 1968, 7, 1517–1521. [Google Scholar] [CrossRef]
- Brown, D.A.; Fanning, M.O.; Fitzpatrick, N.J. Bonding in the (.pi.-(3)-1,2-dicarbollyl)tricarbonylmanganese anion. Inorg. Chem. 1980, 19, 1822–1823. [Google Scholar] [CrossRef]
- Semin, G.K.; Bryukhova, E.V. On the manifestation of electron-nuclear dynamics in 59Co NQR spectra for the series of π-cyclopentadienyl-π-(3)-1,2-dicarbollylcobalt derivatives. Russ. J. Phys. Chem. 2010, 84, 235–239. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Young, D.C.; Andrews, T.D.; Howe, D.V.; Pilling, R.L.; Pitts, A.D.; Reintjer, M.; Warren, L.F.; Wegner, P.A. π-Dicarbollyl derivatives of the transition metals. Metallocene analogs. J. Am. Chem. Soc. 1968, 90, 879–896. [Google Scholar] [CrossRef]
- Grimes, R.N. Transitional metal metallacarbaboranes. In Comprehensive Organometallic Chemistry II; Elsevier: Oxford, UK, 1995; Volume 1, pp. 373–430. [Google Scholar]
- Grimes, R.N. Metallacarboranes in the new millennium. Coord. Chem. Rev. 2000, 200, 773–811. [Google Scholar] [CrossRef]
- Hosmane, N.S.; Maguire, J.A. Metallacarboranes of d- and f-block metals. In Comprehensive Organometallic Chemistry III; Elsevier: Oxford, UK, 2007; Volume 3, pp. 175–264. [Google Scholar]
- Grimes, R.N. Metallacarboranes of the transition and lanthanide elements. In Carboranes, 3rd ed.; Academic Press: London, UK, 2016; pp. 711–903. [Google Scholar] [CrossRef]
- Kar, S.; Pradhan, A.N.; Ghosh, S. Polyhedral metallaboranes and metallacarboranes. In Comprehensive Organometallic Chemistry IV; Elsevier: Oxford, UK, 2022; Volume 9, pp. 263–369. [Google Scholar] [CrossRef]
- Grishin, I.D.; Turmina, E.S.; D’yachihin, D.I.; Peregudova, S.M.; Chizhevsky, I.T.; Grishin, D.F. Ruthenium carborane complexes: A relationship between the structure, electrochemical properties, and reactivity in catalysis of polymerization processes. Russ. Chem. Bull. 2013, 62, 692–698. [Google Scholar] [CrossRef]
- Chan, A.P.Y.; Parkinson, J.A.; Rosair, G.M.; Welch, A.J. Bis(phosphine)hydridorhodacarborane derivatives of 1,1′-bis(ortho-carborane) and their catalysis of alkene isomerization and the hydrosilylation of acetophenone. Inorg. Chem. 2020, 59, 2011–2023. [Google Scholar] [CrossRef]
- Zimina, A.M.; Anufriev, S.A.; Derendyaeva, M.A.; Knyazeva, N.A.; Somov, N.V.; Malysheva, Y.B.; Sivaev, I.B.; Grishin, I.D. Ruthenium complexes of 5-MeC2B9-carborane ligand: Synthesis and application in polymerization catalysis. Dokl. Chem. 2021, 498, 97–103. [Google Scholar] [CrossRef]
- Grishin, I.D.; Zimina, A.M.; Anufriev, S.A.; Knyazeva, N.A.; Piskunov, A.V.; Dolgushin, F.M.; Sivaev, I.B. Synthesis and catalytic properties of novel ruthenacarboranes based on nido-[5-Me-7,8-C2B9H10]2− and nido-[5,6-Me2-7,8-C2B9H9]2− dicarbollide ligands. Catalysts 2021, 11, 1409. [Google Scholar] [CrossRef]
- Causey, P.W.; Besanger, T.R.; Valliant, J.F. Synthesis and screening of mono- and di-aryl technetium and rhenium metallocarboranes. A new class of probes for the estrogen receptor. J. Med. Chem. 2008, 51, 2833–2844. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.; Valliant, J.F. Metallocarborane-based bisphosphonates for tumour imaging and therapy. Nucl. Med. Biol. 2010, 37, 681–682. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, W.; Chen, Y.; Jiang, H.; Yan, H.; Kosenko, I.; Chekulaeva, L.; Sivaev, I.; Bregadze, V.; Wang, X. A Highly potent antibacterial agent targeting methicillin-resistant Staphylococcus aureus based on cobalt bis(1,2-dicarbollide) alkoxy derivative. Organometallics 2017, 36, 3484–3490. [Google Scholar] [CrossRef]
- Gozzi, M.; Schwarze, B.; Hey-Hawkins, E. Half- and mixed-sandwich metallacarboranes for potential applications in medicine. Pure Appl. Chem. 2019, 91, 563–573. [Google Scholar] [CrossRef]
- Gozzi, M.; Murganic, B.; Drača, D.; Popp, J.; Coburger, P.; Maksimović-Ivanić, D.; Mijatović, S.; Hey-Hawkins, E. Quinoline-conjugated ruthenacarboranes: Toward hybrid drugs with a dual mode of action. ChemMedChem 2019, 14, 2061–2074. [Google Scholar] [CrossRef] [Green Version]
- Schwarze, B.; Jelača, S.; Welcke, L.; Maksimović-Ivanić, D.; Mijatović, S.; Hey-Hawkins, E. 2,2′-Bipyridine-modified tamoxifen: A versatile vector for molybdacarboranes. ChemMedChem 2019, 14, 2075–2083. [Google Scholar] [CrossRef] [Green Version]
- Grüner, B.; Brynda, J.; Das, V.; Šícha, V.; Štěpánková, J.; Nekvinda, J.; Holub, J.; Pospíšilová, K.; Fábry, M.; Pachl, P.; et al. Metallacarborane sulfamides: Unconventional, specific, and highly selective inhibitors of carbonic anhydrase IX. J. Med. Chem. 2019, 62, 9560–9575. [Google Scholar] [CrossRef]
- Kellert, M.; Sárosi, I.; Rajaratnam, R.; Meggers, E.; Lönnecke, P.; Hey-Hawkins, E. Ruthenacarborane-phenanthroline derivatives as potential metallodrugs. Molecules 2020, 25, 2322. [Google Scholar] [CrossRef]
- Teixeira, R.G.; Marques, F.; Robalo, M.P.; Fontrodona, X.; Garcia, H.; Geninatti Crich, S.; Viñas, C.; Valente, A. Ruthenium carboranyl complexes with 2,2′-bipyridine derivatives for potential bimodal therapy application. RSC Adv. 2020, 10, 16266–16276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drača, D.; Marković, M.; Gozzi, M.; Mijatović, S.; Maksimović-Ivanić, D.; Hey-Hawkins, E. Ruthenacarborane and quinoline: A promising combination for the treatment of brain tumors. Molecules 2021, 26, 3801. [Google Scholar] [CrossRef] [PubMed]
- Swietnicki, W.; Goldeman, W.; Psurski, M.; Nasulewicz-Goldeman, A.; Boguszewska-Czubara, A.; Drab, M.; Sycz, J.; Goszczyński, T.M. Metallacarborane derivatives effective against Pseudomonas aeruginosa and Yersinia enterocolitica. Int. J. Mol. Sci. 2021, 22, 6762. [Google Scholar] [CrossRef]
- Kubiński, K.; Masłyk, M.; Janeczko, M.; Goldeman, W.; Nasulewicz-Goldeman, A.; Psurski, M.; Martyna, A.; Boguszewska-Czubara, A.; Cebula, J.; Goszczyński, T.M. Metallacarborane derivatives as innovative anti-Candida albicans agents. J. Med. Chem. 2022, 65, 13935–13945. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Chemistry of cobalt bis(dicarbollides). A review. Collect. Czech. Chem. Commun. 1999, 64, 783–805. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Swain, B.R.; Mahanta, C.S.; Jena, B.B.; Hosmane, N.S. Cobalt bis(dicarbollide) anion and its derivatives. J. Organomet. Chem. 2017, 849–850, 170–194. [Google Scholar] [CrossRef]
- Druzina, A.A.; Shmalko, A.V.; Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives of cobalt and iron bis(dicarbollide)s and their use in organic synthesis. Russ. Chem. Rev. 2021, 90, 785–830. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Nelyubina, Y.V.; Pavlov, A.P.; Novikov, V.V.; Shvydkiy, N.V.; Kudinov, A.R. Polyhedral rearrangements in the complexes of rhodium and iridium with isomeric carborane anions [7,8-Me2-X-SMe2-7,8-nido- C2B9H8]− (X = 9 and 10). Organometallics 2017, 36, 791–800. [Google Scholar] [CrossRef]
- Timofeev, S.V.; Zhidkova, O.B.; Sivaev, I.B.; Starikova, Z.A.; Suponitsky, K.Y.; Yan, H.; Bregadze, V.I. Synthesis of rhodacarboranes containing σ- and π-carboranyl ligands in one molecule. J. Organomet. Chem. 2018, 867, 342–346. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Nelyubina, Y.V.; Ikonnikov, N.S. Different reactivity of cyclooctadiene complexes 3,3-(cod)- 8-SMe2-closo-3,1,2-RhC2B9H10 and 1,8-Me2-2,2-(cod)-11-SMe2-2,1,8-closo-RhC2B9H8 toward iodine. J. Organomet. Chem. 2018, 867, 224–227. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Loginov, D.A. Rhoda- and iridacarborane halide complexes: Synthesis, structure and application in homogeneous catalysis. J. Organomet. Chem. 2020, 910, 121135. [Google Scholar] [CrossRef]
- Timofeev, S.V.; Zhidkova, O.B.; Suponitsky, K.Y.; Anisimov, A.A.; Sivaev, I.B.; Yan, H.; Bregadze, V.I. Rhodacarboranes containing σ- and π-carborane ligands. New aspects. Inorg. Chim. Acta 2021, 518, 120243. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I. Chemistry of nickel and iron bis(dicarbollides). A review. J. Organomet. Chem. 2002, 614–615, 27–36. [Google Scholar] [CrossRef]
- Miller, S.B.; Hawthorne, M.F. Novel ligand rearrangement of closo-nickelacarbaboranes. J. Chem. Soc. Chem. Commun. 1976, 19, 786–787. [Google Scholar] [CrossRef]
- King III, R.E.; Miller, S.B.; Knobler, C.B.; Hawthorne, M.F. Simultaneous conversion of NiPR3 and BH to NiH and BPR3 linkages by thermal rearrangement of d8 closo-bis(triarylphosphine)nickelacarboranes. Crystal and molecular structure of [closo-3-(μ-CO)-8-PPh3-3,1,2-NiC2B9H10]2: A dimeric nickelacarborane complex containing a metal-metal bond. Inorg. Chem. 1983, 22, 3548–3554. [Google Scholar] [CrossRef]
- Erdman, A.A.; Zubreichuk, Z.P.; Knizhnikov, V.A.; Maier, A.A.; Aleksandrov, G.G.; Nefedov, S.E.; Eremenko, I.L. Synthesis and the structure of the triphenylphosphine complex of o-nickelacarborane, 3,3-(PPh3)2-3,1,2-NiC2B9H11. Russ. Chem. Bull. 2001, 50, 2248–2250. [Google Scholar] [CrossRef]
- Hodson, B.E.; McGrath, T.D.; Stone, F.G.A. Supraicosahedral and icosahedral nickelacarbaboranes bearing exopolyhedral metal fragments. Dalton Trans. 2004, 16, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Hodson, B.E.; McGrath, T.D.; Stone, F.G.A. Synthesis, structure, and dynamics of nickelacarboranes incorporating the [nido-7,9-C2B9H11]2- ligand. Inorg. Chem. 2004, 43, 3090–3097. [Google Scholar] [CrossRef]
- Wang, L.; Perveen, S.; Ouyang, Y.; Zhang, S.; Jiao, J.; He, G.; Nie, Y.; Li, P. Well-defined, versatile and recyclable half-sandwich nickelacarborane catalyst for selective carbene-transfer reactions. Chem. Eur. J. 2021, 27, 5754–5760. [Google Scholar] [CrossRef]
- Carr, N.; Mullica, D.F.; Sappenfield, E.L.; Stone, F.G.A. Carborane complexes of nickel and platinum: Synthesis and protonation reactions of anionic allyl(carborane) species. Inorg. Chem. 1994, 33, 1666–1673. [Google Scholar] [CrossRef]
- Garrioch, R.M.; Kuballa, P.; Low, K.S.; Rosair, G.M.; Welch, A.J. The ‘1,2 to 1,2’ cage carbon isomerisation in bisphosphine carbanickelaboranes: Synthesis and spectroscopic and crystallographic characterisation of 1,2-Ph2- 4,4-(PMe2Ph)2-4,1,2-closo-NiC2B9H9, 1,2-Ph2-4,4-(PEt3)2-4,1,2-closo-NiC2B9H9 and 1,2-Ph2-4-(dppe)-4,1,2-closo-NiC2B9H9. J. Organomet. Chem. 1999, 575, 57–62. [Google Scholar] [CrossRef]
- Kudinov, A.R.; Petrovskii, P.V.; Meshcheryakov, V.I.; Rybinskaya, M.I. Synthesis of the first metallacarborane triple-decker complexes with a central cyclopentadienyl ligand. Russ. Chem. Bull. 1999, 48, 1356–1361. [Google Scholar] [CrossRef]
- Park, J.-S.; Kim, D.-H.; Kim, S.-J.; Ko, J.; Kim, S.H.; Cho, S.; Lee, C.-H.; Kang, S.O. Preparation and reactions of a half-sandwich dicarbollyl nickel(II) complex containing a dimethylamino pendent group. Organometallics 2001, 20, 4483–4491. [Google Scholar] [CrossRef]
- Robertson, S.; Ellis, D.; Rosair, G.M.; Welch, A.J. Nickelation of [3-Et-7,8-Ph2-7,8-nido-C2B9H8]2−: Synthesis and characterization of 1,2 → 1,2 and 1,2 → 1,7 isomerized products. Appl. Organometal. Chem. 2003, 17, 518–524. [Google Scholar] [CrossRef]
- Robertson, S.; Garrioch, R.M.; Ellis, D.; McGrath, T.D.; Hodson, B.E.; Rosair, G.M.; Welch, A.J. Towards the mechanism of heteroborane isomerisation: 1,2 → 1,2 and 1,2 → 1,7 low-temperature isomerisations from metallations of [5-I-7,8-Ph2-7,8-nido-C2B9H8]2−. Inorg. Chim. Acta 2005, 358, 1485–1493. [Google Scholar] [CrossRef]
- Yinghuai, Z.; Sia, S.L.P.; Carpenter, K.; Kooli, F.; Kemp, R.A. Syntheses and catalytic activities of single-wall carbon nanotubes-supported nickel (II) metallacarboranes for olefin polymerization. J. Phys. Chem. Solids 2006, 67, 1218–1222. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Erokhina, S.A.; Suponitsky, K.Y.; Markov, V.Y.; Sivaev, I.B. Synthesis and crystal structures of nickel(II) and palladium(II) complexes with o-carboranyl amidine ligands. Dalton Trans. 2021, 50, 4967–4975. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Erokhina, S.A.; Suponitsky, K.Y.; Sivaev, I.B.; Bregadze, V.I. Coordination ability of 10-EtC(NHPr)=HN-7,8-C2B9H11 in the reactions with nickel(II) phosphine complexes. Crystals 2021, 11, 306. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Anufriev, S.A.; Sivaev, I.B. Charge-compensated derivatives of nido-carborane. Inorganics 2023, 11, 72. [Google Scholar] [CrossRef]
- Timofeev, S.V.; Sivaev, I.B.; Prikaznova, E.A.; Bregadze, V.I. Transition metal complexes with charge-compensated dicarbollide ligands. J. Organomet. Chem. 2014, 751, 221–250. [Google Scholar] [CrossRef]
- Zhang, D.; Dou, J.; Li, D.; Wang, D. Synthesis and characterization of three group 10 complexes containing nido-carborane diphosphine: [MCl(PPh3){7,8-(PPh2)2-7,8-C2B9H10}] (M = Ni, Pd, Pt). Inorg. Chim. Acta 2006, 13, 4243–4249. [Google Scholar] [CrossRef]
- Axtell, J.C.; Kirlikovali, K.O.; Djurovich, P.I.; Jung, D.; Nguyen, V.T.; Munekiyo, B.; Royappa, A.T.; Rheingold, A.L.; Spokoyny, A.M. Blue phosphorescent zwitterionic iridium(III) complexes featuring weakly coordinating nido-carborane-based ligands. J. Am. Chem. Soc. 2016, 138, 15758–15765. [Google Scholar] [CrossRef]
- Uemura, K.; Tanaka, K.; Chujo, Y. Conformation-dependent electron donation of nido-carborane substituents and its influence on phosphorescence of tris(2,2′-bipyridyl)ruthenium(II) complex. Crystals 2022, 12, 688. [Google Scholar] [CrossRef]
- Yinghuai, Z.; Yulin, Z.; Carpenter, K.; Maguire, J.A.; Hosmane, N.S. Synthesis and catalytic activities of Group 4 metal complexes derived from C(cage)-appended cyclohexyloxocarborane trianion. J. Organomet. Chem. 2005, 690, 2802–2808. [Google Scholar] [CrossRef]
- Yinghuai, Z.; Lo Pei Sia, S.; Kooli, F.; Carpenter, K.; Kemp, R.A. Another example of carborane based trianionic ligand: Syntheses and catalytic activities of cyclohexylamino tailed ortho-carboranyl zirconium and titanium dicarbollides. J. Organomet. Chem. 2005, 690, 6284–6291. [Google Scholar] [CrossRef]
- Shen, H.; Chan, H.-S.; Xie, Z. Guanylation of amines catalyzed by a half-sandwich titanacarborane amide complex. Organometallics 2006, 25, 5515–5517. [Google Scholar] [CrossRef]
- Gao, M.; Tang, Y.; Xie, M.; Qian, C.; Xie, Z. Synthesis, structure, and olefin polymerization behavior of constrained-geometry Group 4 metallacarboranes incorporating imido-dicarbollyl ligands. Organometallics 2006, 25, 2578–2584. [Google Scholar] [CrossRef]
- Shen, H.; Chan, H.-S.; Xie, Z. Synthesis, structure, and reactivity of [σ:η1:η5-(OCH2)(Me2NCH2)C2B9H9]Ti(NR2) (R = Me, Et). Organometallics 2007, 26, 2694–2704. [Google Scholar] [CrossRef]
- Lee, J.-D.; Lee, Y.-J.; Son, K.-C.; Han, W.-S.; Cheong, M.; Ko, J.; Kang, S.O. Synthesis, characterization, and reactivity of new types of constrained geometry Group 4 metal complexes derived from picolyl-substituted dicarbollide ligand systems. J. Organomet. Chem. 2007, 692, 5403–5413. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Sivaev, I.B.; Nakamura, H. Two possible ways to combine boron and gadolinium for Gd-quided BNCT. A concept. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 910–917. [Google Scholar] [CrossRef]
- Park, J.-S.; Kim, D.-H.; Ko, J.; Kim, S.H.; Cho, S.; Lee, C.-H.; Kang, S.O. Half-sandwich iron(II) and ruthenium(II) complexes with the dicarbollylamino ligand system. Organometallics 2001, 20, 4632–4640. [Google Scholar] [CrossRef]
- Kim, D.-H.; Won, J.H.; Kim, S.-J.; Ko, J.; Kim, S.H.; Cho, S.; Kang, S.O. Dicarbollide analogues of the constrained-geometry polymerization catalyst. Organometallics 2001, 20, 4298–4300. [Google Scholar] [CrossRef]
- Lee, J.-D.; Lee, Y.-J.; Son, K.-C.; Cheong, M.; Ko, J.; Kang, S.O. New types of constrained geometry Group 4 metal complexes derived from the aminomethyldicarbollyl ligand system: Synthesis and structural characterization of mono-dicarbollylamino and bis-dicarbollylamino Group 4 metal complexes. Organometallics 2007, 26, 3374–3384. [Google Scholar] [CrossRef]
- Mandal, D.; Man, W.Y.; Rosair, G.M.; Welch, A.J. Steric versus electronic factors in metallacarborane isomerisation: Nickelacarboranes with 3,1,2-, 4,1,2- and 2,1,8-NiC2B9 architectures and pendant carborane groups, derived from 1,1′-bis(o-carborane). Dalton Trans. 2016, 45, 15013–15025. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Rosair, G.M. Exploration of bis(nickelation) of 1,1′-bis(o-carborane). Crystals 2021, 11, 16. [Google Scholar] [CrossRef]
- Viñas, C.; Bertran, J.; Gomez, S.; Teixidor, F.; Dozol, J.-F.; Rouquette, H.; Kivekäs, R.; Sillanpää, R. Aromatic substituted metallacarboranes as extractants of 137Cs and 90Sr from nuclear wastes. J. Chem. Soc. Dalton Trans. 1998, 17, 2849–2854. [Google Scholar] [CrossRef]
- Semyonov, D.K.; Slushko, G.K.; Stogniy, M.Y.; Anufriev, S.A.; Godovikov, I.A.; Suponitsky, K.Y.; Bregadze, V.I.; Sivaev, I.B. Interligand interactions in half-sandwich nickelacarboranes with phosphine ligands: Away from skeletal rearrangements. Organometallics 2023, 42. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Sivaev, I.B.; Petrovskii, P.V.; Bregadze, V.I. Synthesis of monosubstituted functional derivatives of carboranes from 1-mercapto-ortho-carborane: 1-HOOC(CH2)nS-1,2-C2B10H11 and [7-HOOC(CH2)nS-7,8-C2B9H11]−(n = 1–4). Dalton Trans. 2010, 39, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Stogniy, M.Y.; Erokhina, S.A.; Druzina, A.A.; Sivaev, I.B.; Bregadze, V.I. Synthesis of novel carboranyl azides and “click” reactions thereof. J. Organomet. Chem. 2019, 904, 121007. [Google Scholar] [CrossRef]
- Standley, E.A.; Smith, S.J.; Müller, P.; Jamison, T.F. A broadly applicable strategy for entry into homogeneous nickel(0) catalysts from air-stable nickel(II) complexes. Organometallics 2014, 33, 2012–2018. [Google Scholar] [CrossRef] [Green Version]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Burlington, UK, 2009. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semyonov, D.K.; Stogniy, M.Y.; Suponitsky, K.Y.; Sivaev, I.B. Half-Sandwich Nickelacarboranes Derived from [7-(MeO(CH2)2S)-7,8-C2B9H11]−. Inorganics 2023, 11, 127. https://doi.org/10.3390/inorganics11030127
Semyonov DK, Stogniy MY, Suponitsky KY, Sivaev IB. Half-Sandwich Nickelacarboranes Derived from [7-(MeO(CH2)2S)-7,8-C2B9H11]−. Inorganics. 2023; 11(3):127. https://doi.org/10.3390/inorganics11030127
Chicago/Turabian StyleSemyonov, Dmitriy K., Marina Yu. Stogniy, Kyrill Yu. Suponitsky, and Igor B. Sivaev. 2023. "Half-Sandwich Nickelacarboranes Derived from [7-(MeO(CH2)2S)-7,8-C2B9H11]−" Inorganics 11, no. 3: 127. https://doi.org/10.3390/inorganics11030127
APA StyleSemyonov, D. K., Stogniy, M. Y., Suponitsky, K. Y., & Sivaev, I. B. (2023). Half-Sandwich Nickelacarboranes Derived from [7-(MeO(CH2)2S)-7,8-C2B9H11]−. Inorganics, 11(3), 127. https://doi.org/10.3390/inorganics11030127