
Periodic Density Functional Theory (PDFT) Simulating Crystal Structures with Microporous CHA Framework: An Accuracy and Efficiency Study


Abstract
:
1. Introduction
2. Results and Discussion
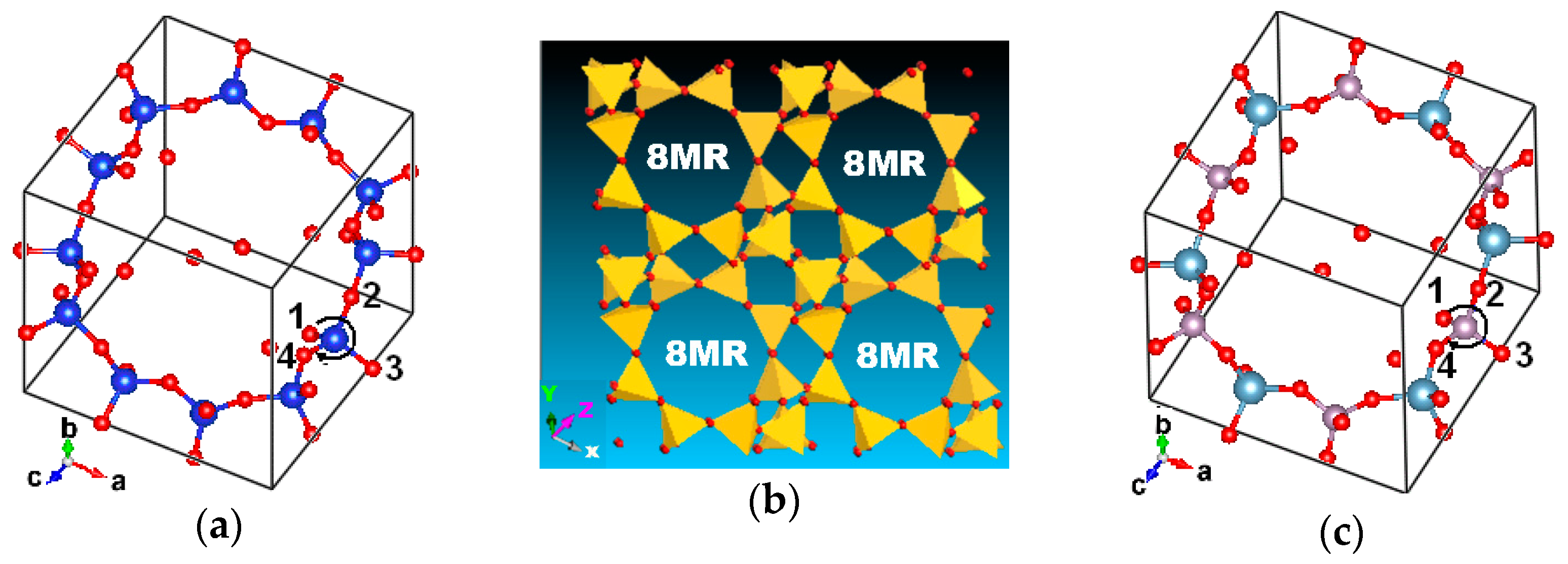
2.1. Topology
2.2. All-Silica Chabazite
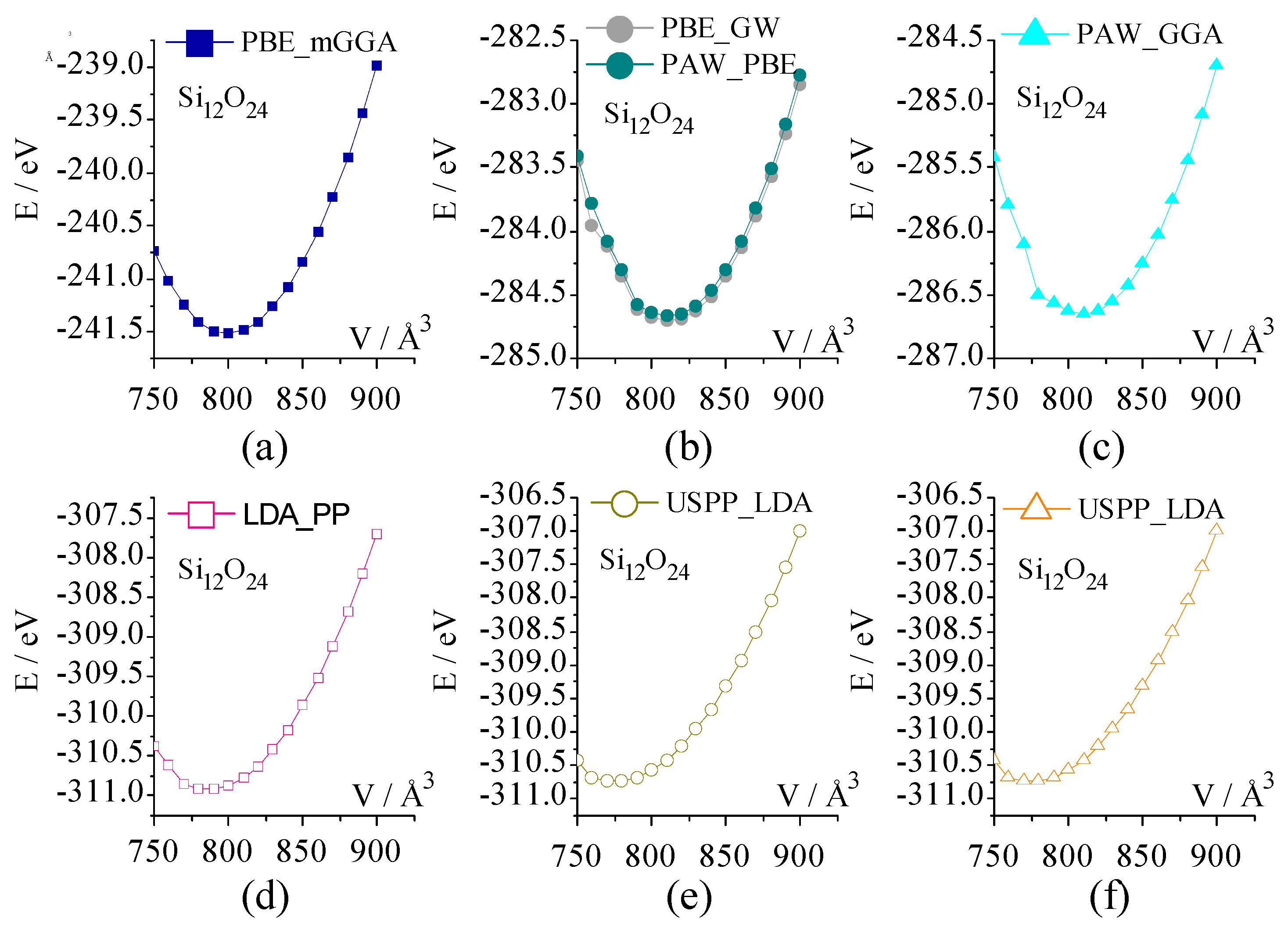
2.2.1. Energy-Volume Curve and Fitted Equilibrium Volume (V0,fitted)
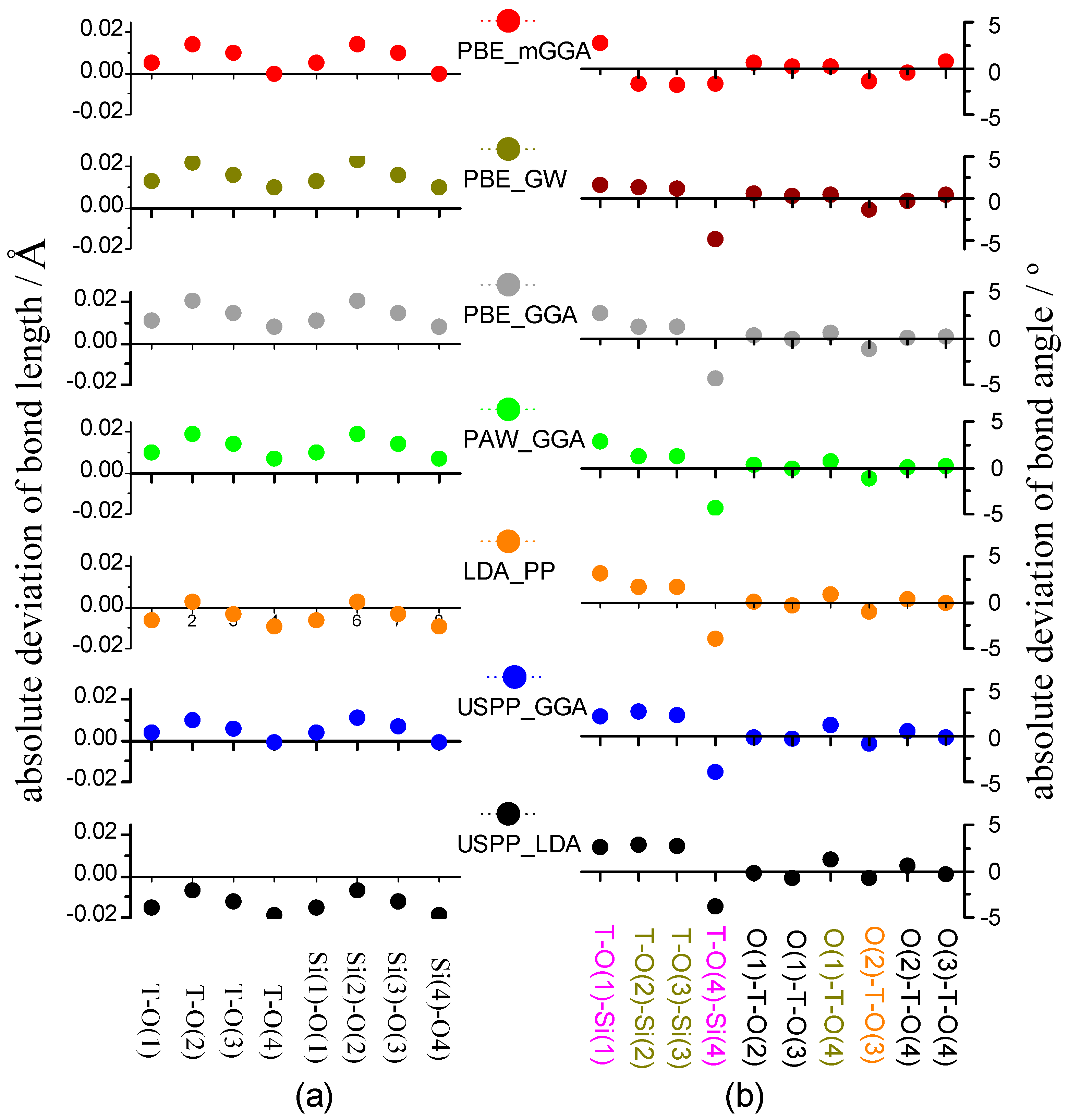
2.2.2. Precise Structure of All-Silica Chabazite and Calculation Error
2.3. AlPO4-34 Framework
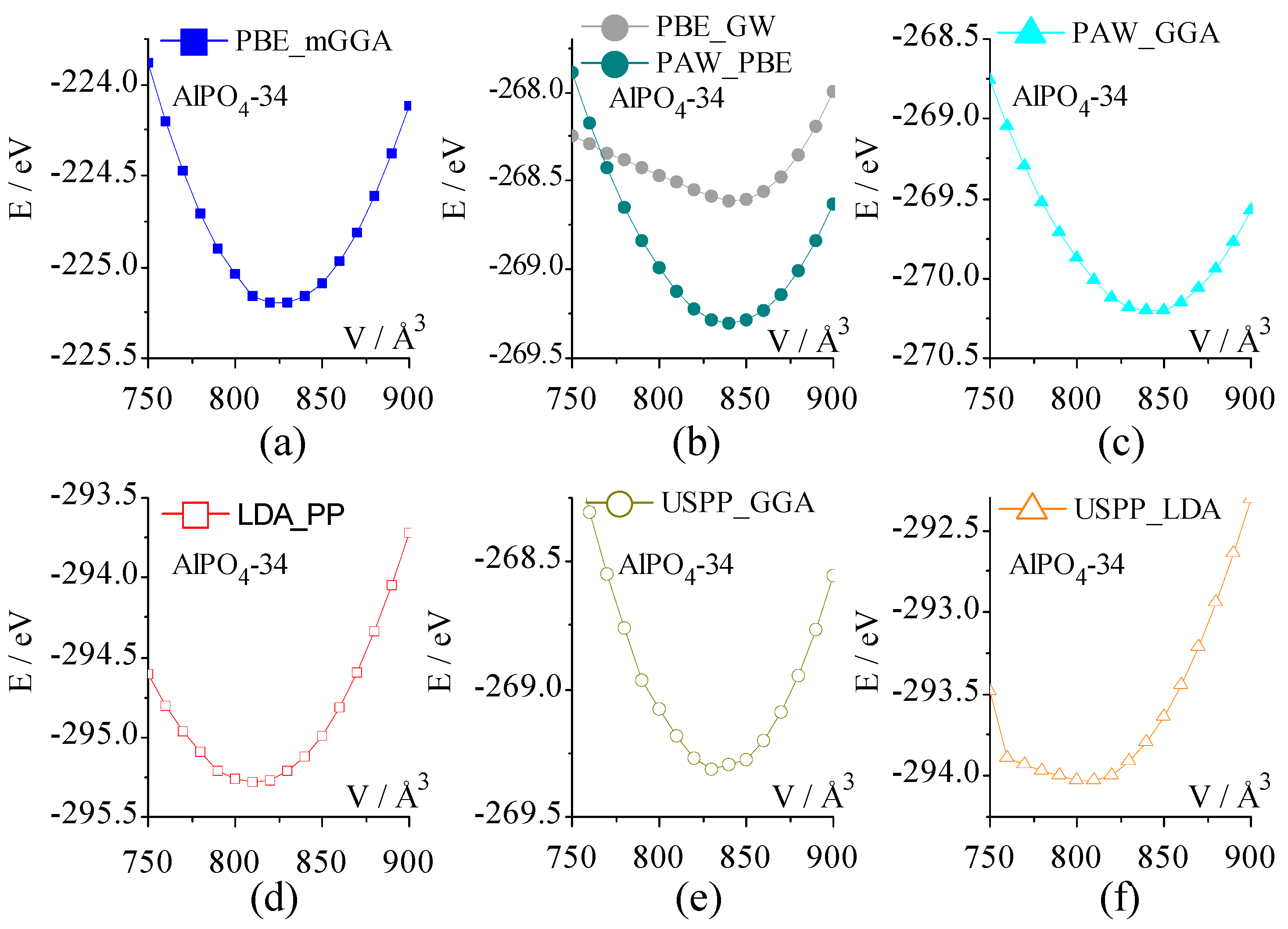
2.3.1. Energy-Volume Curve
2.3.2. Fitted Equilibrium Volume (V0,fitted)

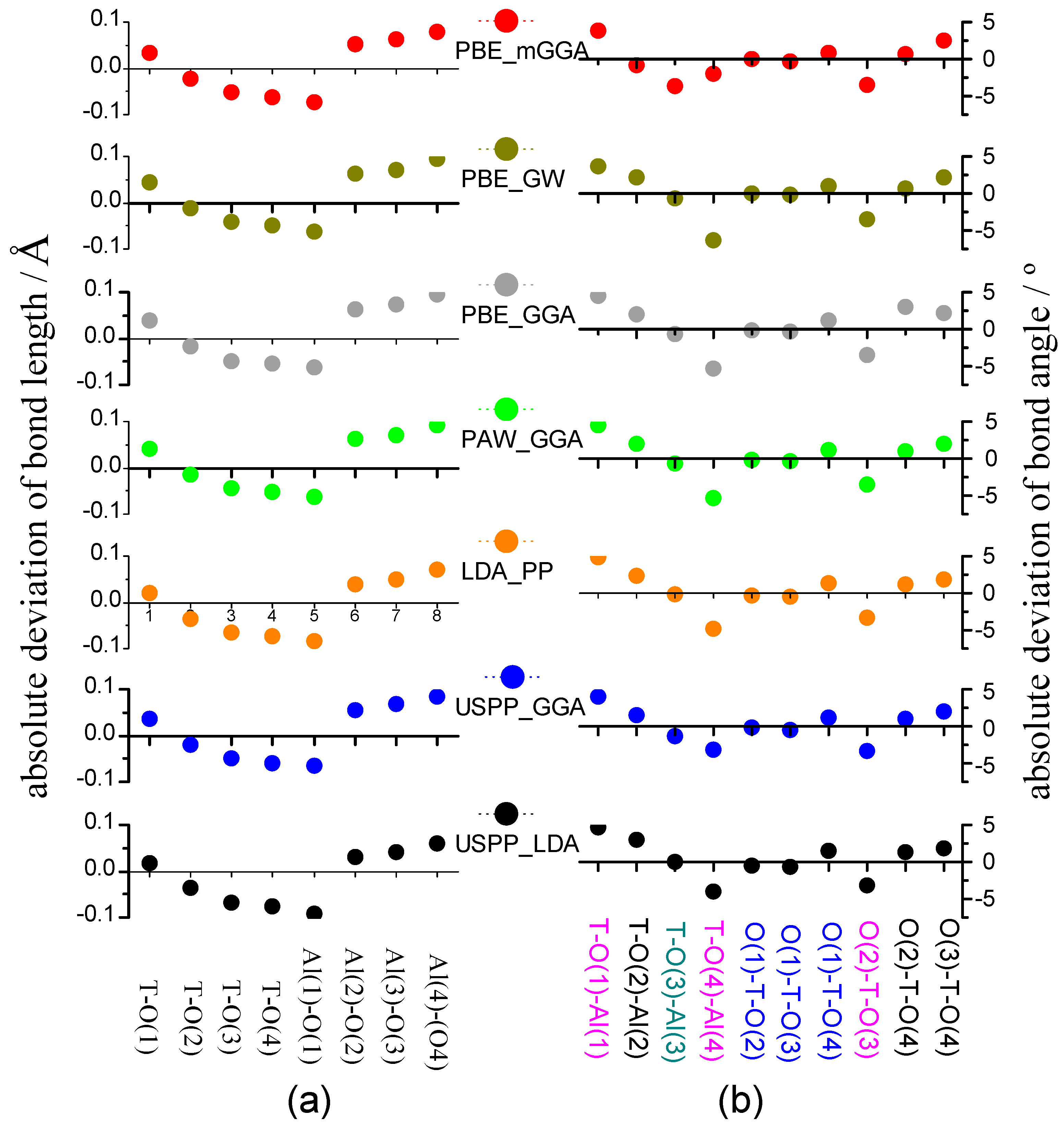
2.3.3. Precise Structure of AlPO4-34 and Calculation Error

2.4. Calculation Time and Phonon Calculation
3. Materials and Methods
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.; Deng, D.; Bao, X. Catalysis for Selected C1 Chemistry. Chem 2020, 6, 2497–2514. [Google Scholar] [CrossRef]
- Tian, P.; Wei, Y.; Ye, M.; Liu, Z. Methanol to Olefins (MTO): From Fundamentals to Commercialization. ACS Catal. 2015, 5, 1922–1938. [Google Scholar] [CrossRef]
- Rimaz, S.; Kosari, M.; Zarinejad, M.; Ramakrishna, S. A Comprehensive Review on Sustainability-Motivated Applications of SAPO-34 Molecular Sieve. J. Mater. Sci. 2022, 57, 848–886. [Google Scholar] [CrossRef]
- Hessou, E.P.; Ponce-Vargas, M.; Mensah, J.-B.; Tielens, F.; Santos, J.C.; Badawi, M. Dibenzyl Disulfide Adsorption on Cationic Exchanged Faujasites: A DFT Study. Nanomaterials 2019, 9, 715. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, X.; Liu, S.; Xie, S.; Zhu, X.; Chen, F.; Xu, L. Activity Enhancement of ZSM-35 in Dimethyl Ether Carbonylation Reaction through Alkaline Modifications. RSC Adv. 2013, 3, 16549–16557. [Google Scholar] [CrossRef]
- Liu, J.; Xue, H.; Huang, X.; Li, Y.; Shen, W. Dimethyl Ether Carbonylation to Methyl Acetate over HZSM-35. Catal. Lett. 2010, 139, 33–37. [Google Scholar] [CrossRef]
- Kim, J.J.; Jeong, D.J.; Jung, H.S.; Hur, Y.G.; Choung, J.W.; Baik, J.H.; Park, M.-J.; Chung, C.-H.; Bae, J.W. Dimethyl ether conversion to hydrocarbons on the closely interconnected FER@ZSM-5 nanostructures. Micropor. Mesopor. Mater. 2022, 340, 112034. [Google Scholar] [CrossRef]
- Li, X.; Chen, X.; Yang, Z.; Zhu, X.; Xu, S.; Xie, S.; Liu, S.; Liu, X.; Xu, L. Seed-assisted synthesis of FER/MOR composite zeolite and its specific catalytic application in carbonylation reaction. Micropor. Mesopor. Mater. 2018, 257, 79–84. [Google Scholar] [CrossRef]
- Xiong, Z.; Zhan, E.; Li, M.; Shen, W. DME carbonylation over a HSUZ-4 zeolite. Chem. Commun. 2020, 56, 3401–3404. [Google Scholar] [CrossRef]
- Shin, J.; Jo, D.; Hong, S.B. Rediscovery of the Importance of Inorganic Synthesis Parameters in the Search for New Zeolites. Acc. Chem. Res. 2019, 52, 1419–1427. [Google Scholar] [CrossRef]
- Xing, R.; Liu, N.; Liu, Y.; Wu, H.W.; Jiang, Y.W.; Chen, L.; He, M.; Wu, P. Novel Solid Acid Catalysts: Sulfonic Acid Group-Functionalized Mesostructured Polymers. Adv. Funct. Mater. 2007, 17, 2455–2461. [Google Scholar] [CrossRef]
- Li, Y.; Yu, J. New Stories of Zeolite Structures: Their Descriptions, Determinations, Predictions, and Evaluations. Chem. Rev. 2014, 114, 7268–7316. [Google Scholar] [CrossRef]
- Opanasenko, M.V.; Rothab, W.J.; Čejka, J. Two-Dimensional Zeolites in Catalysis: Current Status and Perspectives. Catal. Sci. Technol. 2016, 6, 2467. [Google Scholar] [CrossRef]
- Li, J.; Corma, A.; Yu, J. Synthesis of new zeolite structures. Chem. Soc. Rev. 2015, 44, 7112–7127. [Google Scholar] [CrossRef]
- Yu, J.; Xu, R. Rich Structure Chemistry in the Aluminophosphate Family. Acc. Chem. Res. 2003, 36, 481–490. [Google Scholar] [CrossRef]
- Pastore, H.O.; Coluccia, S.; Marchese, L. Porous Aluminophosphates: From Molecular Sieves to Designed Acid Catalysts. Annu. Rev. Mater. Res. 2005, 35, 351–395. [Google Scholar] [CrossRef]
- Trachta, M.; Bulánek, R.; Bludský, O.; Rubeš, M. Brønsted Acidity in Zeolites Measured by Deprotonation Energy. Sci. Rep. 2022, 12, 7301. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.R.; Alvarez, F.; Henriques, C.; Lemos, F.; Lopes, J.M.; Ribeiro, M.F. Review: Structure-Activity Relationship in Zeolites. J. Mol. Catal. A Chem. 1995, 96, 245–270. [Google Scholar]
- Guo, P.; Yan, N.; Wang, L.; Zou, X. Database Mining of Zeolite Structures. Cryst. Growth Des. 2017, 17, 6821–6835. [Google Scholar] [CrossRef]
- Terasaki, O. Study of the Fine Structure of Zeolites and Materials Confined in Zeolites. Acta Chem. Sand. 1991, 45, 785–790. [Google Scholar] [CrossRef]
- Wang, P.; Sun, Q. Synthesis and Characterisation of Zeolite LTA with Sheet Structure. Micro Nano Lett. 2020, 15, 433–436. [Google Scholar] [CrossRef]
- Asano, N.; Asahina, S.; Lu, J.; Xu, J.; Shen, Y.; Qin, Z.; Mintova, S. Advanced Scanning Electron Microscopy Techniques for Structural Characterization of Zeolites. Inorg. Chem. Front. 2022, 9, 4225–4231. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, M.; Wang, L.; Han, J.; Lou, C.; Xu, S.; Zhang, Y.; Wu, R.a.; Tian, P.; Liu, Z. Recognizing the Minimum Structural Units Driving the Crystallization of SAPO-34 in a Top-Down Process. Chem. Eur. J. 2023, 29, e202203886. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hua, Y.; Wang, J.; Dong, X.; Tian, Q.; Han, Y. Recent Progress in the Direct Synthesis of Hierarchical Zeolites: Synthetic Strategies and Characterization Methods. Mater. Chem. Front. 2017, 1, 2195. [Google Scholar] [CrossRef]
- Skylaris, C.-K. A Benchmark for Materials Simulation-Material Properties Can Now Be Predicted Reliably from First-Principles Calculations. Science 2016, 351, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- First, E.L.; Gounaris, C.E.; Wei, J.; Floudas, C.A. Computational Characterization of Zeolite Porous Networks: An Automated Approach. Phys. Chem. Chem. Phys. 2011, 13, 17339–17358. [Google Scholar] [CrossRef] [PubMed]
- van Santen, R.A.; de Man, A.J.M.; Jacobs, W.P.J.H.; Teunissen, E.H.; Kramer, G.J. Lattice Relaxation of Zeolites. Catal. Lett. 1991, 9, 273–286. [Google Scholar] [CrossRef]
- Falcioni, M.; Deem, M.W. A Biased Monte Carlo Scheme for Zeolite Structure Solution. J. Chem. Phys. 1998, 110, 1754. [Google Scholar] [CrossRef]
- Yang, S.; Lach-hab, M.; Vaisman, I.I.; Blaisten-Barojas, E. Identifying Zeolite Frameworks with a Machine Learning Approach. J. Phys. Chem. C 2009, 113, 21721–21725. [Google Scholar] [CrossRef]
- Lu, J.-R.; Li, Y.; Yu, J.-H.; Lu, Y. Predicting Hypothetical Zeolite Frameworks Using Program FraGen. Acta Phys.-Chim. Sin. 2013, 29, 1661–1665. [Google Scholar]
- Svelle, S.; Tuma, C.; Rozanska, X.; Kerber, T.; Sauer, J. Quantum Chemical Modeling of Zeolite-Catalyzed Methylation Reactions: Toward Chemical Accuracy for Barriers. J. Am. Chem. Soc. 2009, 131, 816–825. [Google Scholar] [CrossRef]
- Nastase, S.A.F.; O’Malley, A.J.; Catlow, C.R.A.; Logsdail, A.J. Computational QM/MM investigation of the adsorption of MTH active species in H-Y and H-ZSM-5. Phys. Chem. Chem. Phys. 2019, 21, 2639. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Zhang, G.; Chen, X.; Zang, K.; Li, X.; Xu, L. Specific zone within 8-membered ring channel as catalytic center for carbonylation of dimethyl ether and methanol over FER zeolite. Appl. Catal. A Gen. 2018, 557, 119–124. [Google Scholar] [CrossRef]
- Boronat, M.; Martínez, C.; Corma, A. Mechanistic differences between methanol and dimethyl ether carbonylation in side pockets and large channels of mordenite. Phys. Chem. Chem. Phys. 2011, 13, 2603–2612. [Google Scholar] [CrossRef]
- Car, R. Density Functional Theory: Fixing Jacob’s Ladder. Nat. Chem. 2016, 8, 820–821. [Google Scholar] [CrossRef]
- Kohn, W. Nobel Lecture: Electronic Structure of Matter-Wave Functions and Density Functionals. Rev. Mod. Phys. 1999, 71, 1253–1266. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hellmann, H. A New Approximation Method in the Problem of Many Electrons. J. Chem. Phys. 1935, 3, 61. [Google Scholar] [CrossRef]
- For the Review about the Life of Hans Hellmann. Available online: http://www.tc.chemie.uni-siegen.de/hellmann (accessed on 3 April 2023).
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Lin-Chung, P.J. Limitation of the Pseudopotential Method. Phys. Rev. B 1973, 8, 4043–4045. [Google Scholar] [CrossRef]
- Mackrodt, W.C. A Note on An Aspect of Pseudopotential Theory. Theor. Chim. Acta 1973, 30, 119–126. [Google Scholar] [CrossRef]
- Ziesche, P.; Kurth, S.; Perdew, J.P. Density Functionals from LDA to GGA. Comput. Mater. Sci. 1998, 11, 122–127. [Google Scholar] [CrossRef]
- Schwerdtfeger, P. The Pseudopotential Approximation in Electronic Structure Theory. ChemPhysChem 2011, 12, 3143–3155. [Google Scholar] [CrossRef] [PubMed]
- Cangi, A.; Gross, E.K.U.; Burke, K. Potential Functionals Versus Density Functionals. Phys. Rev. A 2013, 88, 062505. [Google Scholar] [CrossRef]
- Zhang, G.-X.; Reilly, A.M.; Tkatchenko, A.; Scheffler, M. Performance of Various Density-Functional Approximations for Cohesive Properties of 64 Bulk Solids. New J. Phys. 2018, 20, 063020. [Google Scholar] [CrossRef]
- Potter, M.E. Down the Microporous Rabbit Hole of Silicoaluminophosphates: Recent Developments on Synthesis, Characterization, and Catalytic Applications. ACS Catal. 2020, 10, 9758–9789. [Google Scholar] [CrossRef]
- Jeanvoine, Y.; Ángyán, J.G.; Kresse, G.; Hafner, J. Brønsted Acid Sites in HSAPO-34 and Chabazite: An Ab Initio Structural Study. J. Phys. Chem. B 1998, 102, 5573–5580. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.-F. A Periodic Density Functional Theory Study on Methanol Adsorption in HSAPO-34 Zeolites. Chem. Phys. Lett. 2021, 771, 138532. [Google Scholar] [CrossRef]
- Ayadi, T.; Lebègue, S.; Badawi, M. Ab Initio Molecular Dynamics Investigation of the Co-adsorption of Iodine Species with CO and H2O in Silver-Exchanged Chabazite. Phys. Chem. Chem. Phys. 2022, 24, 24992–24998. [Google Scholar] [CrossRef]
- Usman, M.; Ghanem, A.S.; Shah, S.N.A.; Garba, M.D.; Khan, M.Y.; Khan, S.; Humayun, M.; Khan, A.L. A Review on SAPO-34 Zeolite Materials for CO2 Capture and Conversion. Chem. Rec. 2022, 22, e202200039. [Google Scholar] [CrossRef]
- Usman, M. Recent Progress of SAPO-34 Zeolite Membranes for CO2 Separation: A Review. Membranes 2022, 12, 507. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Yuan, L.; Li, D.; Chen, Y. Mathematical Model for the Industrial SMTO Reactor with a SAPO-34 Catalyst. ACS Omega 2023, 8, 9630–9643. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, L.; Chen, B.; Yang, S.; Qian, Y. Comprehensive Energy Analysis and Integration of Coal-Based MTO Process. Energy 2021, 214, 119060. [Google Scholar] [CrossRef]
- Wei, X.; Yuan, L.; Li, W.; Chen, S.; Liu, Z.; Cheng, S.; Li, L.; Wang, C. Facile Fabrication of Hierarchical SAPO-34 in Bifunctional Catalyst for Direct Conversion of Syngas into Light Olefins. Catal. Lett. 2022. [Google Scholar] [CrossRef]
- Zhang, L.; Su, J.; Zhou, H.; Liu, S.; Liu, C.; Jiao, W.; Wang, Y. The Promotional Role of Potassium on InZr/SAPO-34 for Syngas to Light Olefins. Ind. Eng. Chem. Res. 2022, 61, 16616–16623. [Google Scholar] [CrossRef]
- Tian, P.; Su, X.; Wang, Y.; Xia, Q.; Zhang, Y.; Fan, D.; Meng, S.; Liu, Z. Phase-Transformation Synthesis of SAPO-34 and a Novel SAPO Molecular Sieve with RHO Framework Type from a SAPO-5 Precursor. Chem. Mater. 2011, 23, 1406–1413. [Google Scholar] [CrossRef]
- Vallace, A.; Kester, G.; Casteel, W.; Lau, G.; Whitley, R.; Coe, C. A Study of Structural Defects in X- and Y-Type Zeolites and Their Effect on Their Transformation to Aluminum-Rich Chabazite. J. Phys. Chem. C 2021, 125, 12848–12856. [Google Scholar] [CrossRef]
- Venna, S.R.; Carreon, M.A. Microwave assisted phase transformation of silicoaluminophosphate zeolite crystals. J. Mater. Chem. 2009, 19, 3138–3140. [Google Scholar] [CrossRef]
- Feng, P.; Chen, X.-F.; Li, X.; Zhao, D.; Xie, S.; Xu, L.; He, G.-Z. The Distribution Analysis on the Proton Siting and the Acid Strength of the Zeolite Ferrierite: A Computational Study. Micropor. Mesopor. Mater. 2017, 239, 354–362. [Google Scholar] [CrossRef]
- Zheng, T.; Liu, H.; He, P.; Zhang, R.; Meng, X.; Xu, C.; Liu, H.; Yue, Y.; Liu, Z. Post Synthesis of Hierarchical SAPO-34 via Citric acid Etching: Mechanism of Selective Desilication. Micropor. Mesopor. Mater. 2022, 335, 111798. [Google Scholar] [CrossRef]
- Smith, L.J.; Davidson, A.; Cheetham, A.K. Aneutron Diffraction and Infrared Spectroscopy Study of the Acid Form of the Aluminosilicate Zeolite, Chabazite (H-SSZ-13). Catal. Lett. 1997, 49, 143–146. [Google Scholar] [CrossRef]
- Smith, L.; Cheetham, A.K.; Marchese, L.; Thomas, J.M.; Wright, P.A.; Chen, J.; Gianotti, E. A Quantitative Description of the Active Sites in the Dehydrated Acid Catalyst HSAPO-34 for the Conversion of Methanol to Olefins. Catal. Lett. 1996, 41, 13–16. [Google Scholar] [CrossRef]
- Smith, L.; Cheetham, A.K.; Morris, R.E.; Marchese, L.; Thomas, J.M.; Wright, P.A.; Chen, J. On the Nature of Water Bound to a Solide Acid Catalyst. Science 1996, 271, 799–802. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B Condens. Matter. Mater. Phys. 1993, 48, 13115–13118. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef]
- Zhang, Y.; Kitchaev, D.A.; Yang, J.; Chen, T.; Dacek, S.T.; Samiento-Pérez, R.A.; Marques, M.A.L.; Peng, H.; Ceder, G.; Perdew, J.P.; et al. Efficient First-Principles Prediction of Solid Stability: Towards Chemical Accuracy. npj Comput. Mater. 2018, 4, 9. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Moleclar-Dynamics Simulation of the Liquid-Metal Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B Condens. Matter. Mater. Phys. 1994, 49, 14251. [Google Scholar] [CrossRef]
- Klimes, J.; Kaltak, M.; Kresse, G. Predictive GW Calculations Using Plane Waves and Pseudopotentials. Phys. Rev. B 2014, 90, 075125. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-wave Method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Baerlocher, C.; McCusker, L.B. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases (accessed on 3 April 2023).
- Flanigen, E.M. SMIII mechanism. Elsevier Sci. B 1986, 85, 653–685. [Google Scholar]
- Zang, K.; Zhang, W.; Huang, J.; Ding, J.; Yan, J. Electronic Structure of Chabazite Zeolites H-SSZ-13 and H-SAPO-34. Micropor. Mesopor. Mater. 2022, 338, 111957. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pseudopotential | a/Å | α/° | V0/Å3 | V0,fitted/Å3 |
|---|---|---|---|---|
| PBE_mGGA | 9.31 | 94.59 | 797.61 | 799.75 |
| PBE_GW | 9.35 | 94.28 | 810.89 | 810.90 |
| PAW_PBE | 9.35 | 94.40 | 810.64 | 811.84 |
| PAW_GGA | 9.35 | 94.34 | 809.12 | 810. 80 |
| LDA_PP | 9.26 | 94.40 | 786.88 | 789.13 |
| USPP_GGA | 9.31 | 94.21 | 800.36 | 801.54 |
| USPP_LDA | 9.21 | 94.18 | 775.43 | 777.39 |
| Exp [62] | 9.28 | 94.27 | 792.32 | - |
| Pseudopotential | Chabazite | AlPO4-34 | ||
|---|---|---|---|---|
| RT-O/Å | A/° | RT-O/Å | A/° | |
| PBE_mGGA | 0.016 | 1.382 | 0.066 | 2.310 |
| PBE_GW | 0.016 | 1.804 | 0.059 | 2.771 |
| PAW_PBE | 0.015 | 1.789 | 0.060 | 2.813 |
| PAW_GGA | 0.013 | 1.794 | 0.059 | 2.658 |
| LDA_PP | 0.006 | 1.824 | 0.059 | 2.664 |
| USPP_GGA | 0.007 | 1.867 | 0.058 | 2.181 |
| USPP_LDA | 0.014 | 1.990 | 0.058 | 2.543 |
| Pseudopotential | a/Å | α/° | V0/Å3 | V0,fitted/Å3 |
|---|---|---|---|---|
| PBE_mGGA | 9.41 | 94.55 | 825.11 | 826.90 |
| PBE_GW | 9.47 | 94.35 | 840.84 | 837.27 |
| PAW_PBE | 9.46 | 94.40 | 839.85 | 840.78 |
| PAW_GGA | 9.47 | 94.40 | 841.39 | 842.27 |
| LDA_PP | 9.36 | 94.40 | 812.03 | 811.05 |
| USPP_GGA | 9.45 | 94.36 | 835.35 | 835.20 |
| USPP_LDA | 9.33 | 94.29 | 806.08 | 800.71 |
| Exp [63] | 9.40 | 94.27 | 822.39 | - |
| Pseudopotentials | CPU Times (Seconds) | |||||
|---|---|---|---|---|---|---|
| All-Silica Chabazite | AlPO4-34 | |||||
| E-V Scan | Full-Opt * | Phonon | E-V Scan | Full-Opt * | Phonon | |
| PBE_mGGA | 31,803 | 553 | 25,980 | 91,328 | 718 | 24,890 |
| PBE_GW | 72,179 | 1449 | 23,842 | 178,857 | 1491 | 19,849 |
| PAW_PBE | 18,862 | 80 | 10,076 | 43,155 | 277 | 10,689 |
| PAW_GGA | 47,542 | 80 | 11,703 | 66,963 | 356 | 11,064 |
| LDA_PP | 15,824 | 310 | 9902 | 37,538 | 237 | 10,133 |
| USPP_GGA | 177,071 | 221 | 11,422 | 401,072 | 14,747 | 10,757 |
| USPP_LDA | 14,390 | 170 | 8438 | 31,803 | 285 | 9965 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.-F. Periodic Density Functional Theory (PDFT) Simulating Crystal Structures with Microporous CHA Framework: An Accuracy and Efficiency Study. Inorganics 2023, 11, 215. https://doi.org/10.3390/inorganics11050215
Chen X-F. Periodic Density Functional Theory (PDFT) Simulating Crystal Structures with Microporous CHA Framework: An Accuracy and Efficiency Study. Inorganics. 2023; 11(5):215. https://doi.org/10.3390/inorganics11050215
Chicago/Turabian StyleChen, Xiao-Fang. 2023. "Periodic Density Functional Theory (PDFT) Simulating Crystal Structures with Microporous CHA Framework: An Accuracy and Efficiency Study" Inorganics 11, no. 5: 215. https://doi.org/10.3390/inorganics11050215
APA StyleChen, X. -F. (2023). Periodic Density Functional Theory (PDFT) Simulating Crystal Structures with Microporous CHA Framework: An Accuracy and Efficiency Study. Inorganics, 11(5), 215. https://doi.org/10.3390/inorganics11050215