
Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes †




Abstract
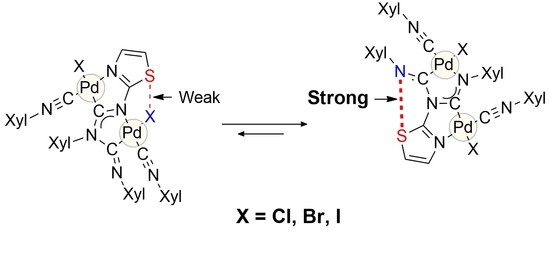
:
1. Introduction
2. Results and Discussions
2.1. Synthesis of Binuclear Diaminocarbene Complexes
2.2. The Regioisomerization
2.3. Characterization of the Regioisomers 4a,b–5a,b
2.4. Analysis of the Intramolecular Chalcogen Bonding
3. Materials and Methods
3.1. General
3.2. Synthesis of the Complexes
3.3. Characterisation of the Complexes






3.4. Data for the Intermediate Complexes 6a and 6b


3.5. X-ray Diffraction
3.6. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the Chalcogen Bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-Hole Concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Jackson, N.E.; Savoie, B.M.; Kohlstedt, K.L.; Olvera de la Cruz, M.; Schatz, G.C.; Chen, L.X.; Ratner, M.A. Controlling Conformations of Conjugated Polymers and Small Molecules: The Role of Nonbonding Interactions. J. Am. Chem. Soc. 2013, 135, 10475–10483. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Gong, Z.; Yan, Q. Chalcogen-Bonding Supramolecular Polymers. J. Org. Chem. 2020, 85, 8397–8404. [Google Scholar] [CrossRef]
- Zeng, R.; Gong, Z.; Chen, L.; Yan, Q. Solution Self-Assembly of Chalcogen-Bonding Polymer Partners. ACS Macro. Lett. 2020, 9, 1102–1107. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef]
- Reid, R.C.; Yau, M.-K.; Singh, R.; Lim, J.; Fairlie, D.P. Stereoelectronic Effects Dictate Molecular Conformation and Biological Function of Heterocyclic Amides. J. Am. Chem. Soc. 2014, 136, 11914–11917. [Google Scholar] [CrossRef]
- Fick, R.J.; Kroner, G.M.; Nepal, B.; Magnani, R.; Horowitz, S.; Houtz, R.L.; Scheiner, S.; Trievel, R.C. Sulfur–Oxygen Chalcogen Bonding Mediates AdoMet Recognition in the Lysine Methyltransferase SET7/9. ACS Chem. Biol. 2016, 11, 748–754. [Google Scholar] [CrossRef]
- Iwaoka, M.; Babe, N. Mining and Structural Characterization of S···X Chalcogen Bonds in Protein Database. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 1257–1264. [Google Scholar] [CrossRef]
- Carugo, O.; Resnati, G.; Metrangolo, P. Chalcogen Bonds Involving Selenium in Protein Structures. ACS Chem. Biol. 2021, 16, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Iwaoka, M. Chalcogen-Containing Protein and Nucleic Acid Derivatives—Synthesis and Applications. In Chalcogen Chemistry: Fundamentals and Applications; The Royal Society of Chemistry: London, UK, 2023; pp. 625–647. [Google Scholar]
- Suresh, K.; Minkov, V.S.; Namila, K.K.; Derevyannikova, E.; Losev, E.; Nangia, A.; Boldyreva, E.V. Novel Synthons in Sulfamethizole Cocrystals: Structure–Property Relations and Solubility. Cryst. Growth Des. 2015, 15, 3498–3510. [Google Scholar] [CrossRef]
- Kumar, V.; Triglav, M.; Morin, V.M.; Bryce, D.L. Predictability of Chalcogen-Bond-Driven Crystal Engineering: An X-Ray Diffraction and Selenium-77 Solid-State NMR Investigation of Benzylic Selenocyanate Cocrystals. ACS Org. Inorg. Au 2022, 2, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, W. Pseudo-Bifurcated Chalcogen Bond in Crystal Engineering. Crystals 2018, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Peloquin, A.J.; McMillen, C.D.; Iacono, S.T.; Pennington, W.T. Crystal Engineering Using Polyiodide Halogen and Chalcogen Bonding to Isolate the Phenothiazinium Radical Cation and Its Rare Dimer, 10-(3-Phenothiazinylidene)Phenothiazinium. Chem. Eur. J. 2021, 27, 8398–8405. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kumar, V.; Bradshaw, M.J.Z.; Bryce, D.L. Chalcogen-Bonded Cocrystals of Substituted Pyridine N-Oxides and Chalcogenodiazoles: An X-ray Diffraction and Solid-State NMR Investigation. Cryst. Growth Des. 2020, 20, 7910–7920. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Elder, P.J.W.; Lee, L.M.; Vargas-Baca, I. The Role of the Lewis Acid−base Properties in the Supramolecular Association of 1,2,5-Chalcogenadiazoles. Can. J. Chem. 2013, 91, 338–347. [Google Scholar] [CrossRef]
- Mikherdov, A.; Novikov, A.; Kinzhalov, M.; Zolotarev, A.; Boyarskiy, V. Intra-/Intermolecular Bifurcated Chalcogen Bonding in Crystal Structure of Thiazole/Thiadiazole Derived Binuclear (Diaminocarbene)PdII Complexes. Crystals 2018, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.C.; Marques, I.; Thompson, A.L.; Christensen, K.E.; Félix, V.; Beer, P.D. Chalcogen Bonding Macrocycles and [2]Rotaxanes for Anion Recognition. J. Am. Chem. Soc. 2017, 139, 3122–3133. [Google Scholar] [CrossRef]
- Wojtkowiak, K.; Michalczyk, M.; Zierkiewicz, W.; Jezierska, A.; Panek, J.J. Chalcogen Bond as a Factor Stabilizing Ligand Conformation in the Binding Pocket of Carbonic Anhydrase IX Receptor Mimic. Int. J. Mol. Sci. 2022, 23, 13701. [Google Scholar] [CrossRef]
- Alfuth, J.; Zadykowicz, B.; Wicher, B.; Kazimierczuk, K.; Połoński, T.; Olszewska, T. Cooperativity of Halogen- and Chalcogen-Bonding Interactions in the Self-Assembly of 4-Iodoethynyl- and 4,7-Bis(Iodoethynyl)Benzo-2,1,3-Chalcogenadiazoles: Crystal Structures, Hirshfeld Surface Analyses, and Crystal Lattice Energy Calculations. Cryst. Growth Des. 2022, 22, 1299–1311. [Google Scholar] [CrossRef]
- Mullin, W.J.; Sharber, S.A.; Thomas, S.W. Optimizing the Self-assembly of Conjugated Polymers and Small Molecules through Structurally Programmed Non-covalent Control. J. Polym. Sci. 2021, 59, 1643–1663. [Google Scholar] [CrossRef]
- Rahman, F.-U.; Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Ballester, P.; Rebek, J.; Yu, Y. Chalcogen Bonding and Hydrophobic Effects Force Molecules into Small Spaces. J. Am. Chem. Soc. 2020, 142, 5876–5883. [Google Scholar] [CrossRef]
- Iwaoka, M.; Isozumi, N. Hypervalent Nonbonded Interactions of a Divalent Sulfur Atom. Implications in Protein Architecture and the Functions. Molecules 2012, 17, 7266–7283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piña, M.d.l.N.; Frontera, A.; Bauza, A. Charge Assisted S/Se Chalcogen Bonds in SAM Riboswitches: A Combined PDB and Ab Initio Study. ACS Chem. Biol. 2021, 16, 1701–1708. [Google Scholar] [CrossRef]
- Pizzi, A.; Daolio, A.; Beccaria, R.; Demitri, N.; Viani, F.; Resnati, G. Chalcogen Bonding (ChB) as a Robust Supramolecular Recognition Motif of Benzisothiazolinone Antibacterials. Chem. Eur. J. 2023, 2023, e202300571. [Google Scholar] [CrossRef]
- Shukla, R.; Dhaka, A.; Aubert, E.; Vijayakumar-Syamala, V.; Jeannin, O.; Fourmigué, M.; Espinosa, E. Understanding Reactivity and Assembly of Dichalcogenides: Structural, Electrostatic Potential, and Topological Analyses of 3H -1,2-Benzodithiol-3-One and Selenium Analogs. Cryst. Growth Des. 2020, 20, 7704–7725. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, M.; Deng, R.; Zheng, P.; Chi, Y.R. Chalcogen Bond-Guided Conformational Isomerization Enables Catalytic Dynamic Kinetic Resolution of Sulfoxides. Nat. Commun. 2022, 13, 4793. [Google Scholar] [CrossRef]
- Gatti, C.; Dessì, A.; Dallocchio, R.; Mamane, V.; Cossu, S.; Weiss, R.; Pale, P.; Aubert, E.; Peluso, P. Factors Impacting σ- and π-Hole Regions as Revealed by the Electrostatic Potential and Its Source Function Reconstruction: The Case of 4,4′-Bipyridine Derivatives. Molecules 2020, 25, 4409. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Halogen Bonding Versuschalcogen and Pnicogen Bonding: A Combined Cambridge Structural Database and Theoretical Study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Carugo, O.I. Chalcogen Bonds Formed by Protein Sulfur Atoms in Proteins. A Survey of High-Resolution Structures Deposited in the Protein Data Bank. J. Biomol. Struct. Dyn. 2022, 91, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Thomas, S.P.; Kottokkaran, R.; Nayak, S.K.; Ramamurthy, P.C.; Guru Row, T.N. A Donor–Acceptor–Donor Structured Organic Conductor with S···S Chalcogen Bonding. Cryst. Growth Des. 2014, 14, 459–466. [Google Scholar] [CrossRef]
- Yasuda, T.; Shimizu, T.; Liu, F.; Ungar, G.; Kato, T. Electro-Functional Octupolar π-Conjugated Columnar Liquid Crystals. J. Am. Chem. Soc. 2011, 133, 13437–13444. [Google Scholar] [CrossRef]
- Kříž, K.; Fanfrlík, J.; Lepšík, M. Chalcogen Bonding in Protein−Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. ChemPhysChem 2018, 19, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Boyarskiy, V.P.; Boyarskaya, I.A.; Dar’in, D.V.; Starova, G.L.; Kukushkin, V.Y. Difference in Energy between Two Distinct Types of Chalcogen Bonds Drives Regioisomerization of Binuclear (Diaminocarbene)PdII Complexes. J. Am. Chem. Soc. 2016, 138, 14129–14137. [Google Scholar] [CrossRef] [PubMed]
- Mikherdov, A.S.; Popov, R.A.; Kinzhalov, M.A.; Haukka, M.; Polukeev, V.A.; Boyarskiy, V.P.; Roodt, A. Reaction Mechanism of Regioisomerization in Binuclear (Diaminocarbene)PdII Complexes. Inorg. Chim. Acta 2021, 514, 120012. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Guedes da Silva, M.F.C.; Resnati, G.; Pombeiro, A.J.L. Chalcogen Bonding in Coordination Chemistry. Coord. Chem. Rev. 2022, 464, 214556. [Google Scholar] [CrossRef]
- Nagao, Y.; Miyamoto, S.; Miyamoto, M.; Takeshige, H.; Hayashi, K.; Sano, S.; Shiro, M.; Yamaguchi, K.; Sei, Y. Highly Stereoselective Asymmetric Pummerer Reactions That Incorporate Intermolecular and Intramolecular Nonbonded S···O Interactions. J. Am. Chem. Soc. 2006, 128, 9722–9729. [Google Scholar] [CrossRef]
- Fukata, Y.; Asano, K.; Matsubara, S. Facile Net Cycloaddition Approach to Optically Active 1,5-Benzothiazepines. J. Am. Chem. Soc. 2015, 137, 5320–5323. [Google Scholar] [CrossRef]
- Abdelhamid, Y.; Kasten, K.; Dunne, J.; Hartley, W.C.; Young, C.M.; Cordes, D.B.; Slawin, A.M.Z.; Ng, S.; Smith, A.D. Isothiourea-Catalyzed [2 + 2] Cycloaddition of C(1)-Ammonium Enolates and N-Alkyl Isatins. Org. Lett. 2022, 24, 5444–5449. [Google Scholar] [CrossRef]
- Zhang, S.; Hartley, W.C.; Greenhalgh, M.D.; Ng, S.; Slawin, A.M.Z.; Smith, A.D. Isothiourea-Catalyzed Synthesis of Pyrrole- and Indole-Functionalized Tetrasubstituted Pyridines. ChemCatChem 2020, 12, 4522–4525. [Google Scholar] [CrossRef]
- Fukumoto, S.; Nakashima, T.; Kawai, T. Photon-Quantitative Reaction of a Dithiazolylarylene in Solution. Angew. Chem. Int. Ed. 2011, 50, 1565–1568. [Google Scholar] [CrossRef]
- Chandrasekhar, V.; Chivers, T.; Ellis, L.; Krouse, I.; Parvez, M.; Vargas-Baca, I. Intramolecular Redox Cyclization upon Oxidation of a Sulfur(II)-Containing Diazene: X-ray Structures of (Ar = 4-CH3C6H4) and MeSO2N(4-CH3C6H4)CN=NC(C6H4CH3-4)NSO2Me. Can. J. Chem. 1997, 75, 1188–1194. [Google Scholar] [CrossRef] [Green Version]
- Akiba, K.; Kobayashi, T.; Arai, S. Chemistry of Hypervalent Sulfur. 9. Bond Switch with Participation of .Pi.-Bonded SIV in a Thiadiazole Ring System. J. Am. Chem. Soc. 1979, 101, 5857–5858. [Google Scholar] [CrossRef]
- Cox, P.A.; Leach, A.G.; Campbell, A.D.; Lloyd-Jones, G.C. Protodeboronation of Heteroaromatic, Vinyl, and Cyclopropyl Boronic Acids: PH–Rate Profiles, Autocatalysis, and Disproportionation. J. Am. Chem. Soc. 2016, 138, 9145–9157. [Google Scholar] [CrossRef] [Green Version]
- Sawwan, N.; Brzostowska, E.M.; Greer, A. Substituent Effects on the Reactivity of Benzo-1,2-Dithiolan-3-One 1-Oxides and Their Possible Application to the Synthesis of DNA-Targeting Drugs. J. Org. Chem. 2005, 70, 6968–6971. [Google Scholar] [CrossRef]
- Ruff, F.; Kapovits, I.; Rábai, J.; Kucsman, Á. Neighbouring Group Participation in Reactions of Sulphides with Chloramine-t. Tetrahedron 1978, 34, 2767–2774. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Timofeeva, S.A.; Luzyanin, K.V.; Boyarskiy, V.P.; Yakimanskiy, A.A.; Haukka, M.; Kukushkin, V.Y. Palladium(II)-Mediated Addition of Benzenediamines to Isocyanides: Generation of Three Types of Diaminocarbene Ligands Depending on the Isomeric Structure of the Nucleophile. Organometallics 2016, 35, 218–228. [Google Scholar] [CrossRef] [Green Version]
- Mikhaylov, V.N.; Sorokoumov, V.N.; Korvinson, K.A.; Novikov, A.S.; Balova, I.A. Synthesis and Simple Immobilization of Palladium(II) Acyclic Diaminocarbene Complexes on Polystyrene Support as Efficient Catalysts for Sonogashira and Suzuki–Miyaura Cross-Coupling. Organometallics 2016, 35, 1684–1697. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths Determined by X-Ray and Neutron-Diffraction. Part 1. Bond Lengths in Organic-Compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Alvarez, S. A Cartography of the van Der Waals Territories. Dalton Trans. 2013, 42, 8617. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular Hydrogen Bond Energies in Crystals Evaluated Using Electron Density Properties: DFT Computations with Periodic Boundary Conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Kashina, M.V.; Mikherdov, A.S.; Mozheeva, E.A.; Novikov, A.S.; Smirnov, A.S.; Ivanov, D.M.; Kryukova, M.A.; Ivanov, A.Y.; Smirnov, S.N.; et al. Dramatically Enhanced Solubility of Halide-Containing Organometallic Species in Diiodomethane: The Role of Solvent⋅⋅⋅Complex Halogen Bonding. Angew. Chem. Int. Ed. 2018, 57, 12785–12789. [Google Scholar] [CrossRef] [PubMed]
- Palatinus, L.; Prathapa, S.J.; van Smaalen, S. EDMA: A Computer Program for Topological Analysis of Discrete Electron Densities. J. Appl. Crystallogr. 2012, 45, 575–580. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta. Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixture | Equilibrium Ratio | Equilibrium Constant | –∆G(CHCl3), kcal/mol |
|---|---|---|---|
| 3a/3b [36] | 13/87 | 6.7 | 1.2 |
| 4a/4b | 4/96 | 24 | 2.0 |
| 5a/5b | 4/96 | 24 | 2.0 |
| Complex | 4b | 5b |
|---|---|---|
| d(Pd1–Ccarbene) | 2.014(2) | 2.034(3) |
| d(N2–Ccarbene) | 1.407(3) | 1.409(4) |
| d(N3–Ccarbene) | 1.286(3) | 1.290(4) |
| d(Pd2–Ccarbene2) | 1.986(2) | 2.007(3) |
| d(N2–Ccarbene2) | 1.438(3) | 1.444(4) |
| d(N4–Ccarbene2) | 1.265(3) | 1.259(4) |
| ∠(N1–Pd1–Br1) | 94.94(6) | |
| ∠(N3–Pd2–Br2) | 97.05(6) | |
| ∠(N1–Pd1–I1) | 95.95(8) | |
| ∠(N3–Pd2–I2) | 97.73(7) |
| Compound | Contact | ρ(r) | ∇2ρ(r) | Hb | V(r) | G(r) | Eint a | Eint b | l |
|---|---|---|---|---|---|---|---|---|---|
| Solid-state structures | |||||||||
| 3b c | S···N | 0.026 | 0.080 | 0.001 | −0.019 | 0.019 | 6.0 | 5.1 | 2.67 |
| 4b | S···N | 0.024 | 0.076 | 0.001 | −0.017 | 0.018 | 5.3 | 4.9 | 2.71 |
| 5b | S···N | 0.024 | 0.076 | 0.001 | −0.017 | 0.018 | 5.3 | 4.9 | 2.70 |
| Optimized structures (CHCl3) | |||||||||
| 3a c | S···Cl | 0.015 | 0.051 | 0.002 | −0.009 | 0.011 | 3.0 | 2.8 | 3.14 |
| 3b c | S···N | 0.024 | 0.071 | 0.001 | −0.016 | 0.017 | 5.0 | 4.6 | 2.73 |
| 4a | S···Br | 0.016 | 0.049 | 0.002 | −0.009 | 0.011 | 2.8 | 3.0 | 3.22 |
| 4b | S···N | 0.024 | 0.073 | 0.001 | −0.017 | 0.018 | 5.3 | 4.9 | 2.72 |
| 5a | S···I | 0.011 | 0.031 | 0.001 | −0.005 | 0.006 | 1.6 | 1.6 | 3.58 |
| 5b | S···N | 0.025 | 0.074 | 0.001 | −0.017 | 0.018 | 5.3 | 4.9 | 2.71 |
| System | −∆Gexp | −∆Gcalcd |
|---|---|---|
| 3a –> 3b * | 1.2 | 3.2 |
| 4a –> 4b | 2.0 | 5.4 |
| 5a –> 5b | 2.0 | 5.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popov, R.A.; Novikov, A.S.; Suslonov, V.V.; Boyarskiy, V.P. Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes. Inorganics 2023, 11, 255. https://doi.org/10.3390/inorganics11060255
Popov RA, Novikov AS, Suslonov VV, Boyarskiy VP. Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes. Inorganics. 2023; 11(6):255. https://doi.org/10.3390/inorganics11060255
Chicago/Turabian StylePopov, Roman A., Alexander S. Novikov, Vitalii V. Suslonov, and Vadim P. Boyarskiy. 2023. "Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes" Inorganics 11, no. 6: 255. https://doi.org/10.3390/inorganics11060255
APA StylePopov, R. A., Novikov, A. S., Suslonov, V. V., & Boyarskiy, V. P. (2023). Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes. Inorganics, 11(6), 255. https://doi.org/10.3390/inorganics11060255