
Turning-On of Coumarin Phosphorescence in Acetylacetonato Platinum Complexes of Cyclometalated Pyridyl-Substituted Coumarins


Abstract
:1. Introduction
2. Results and Discussion
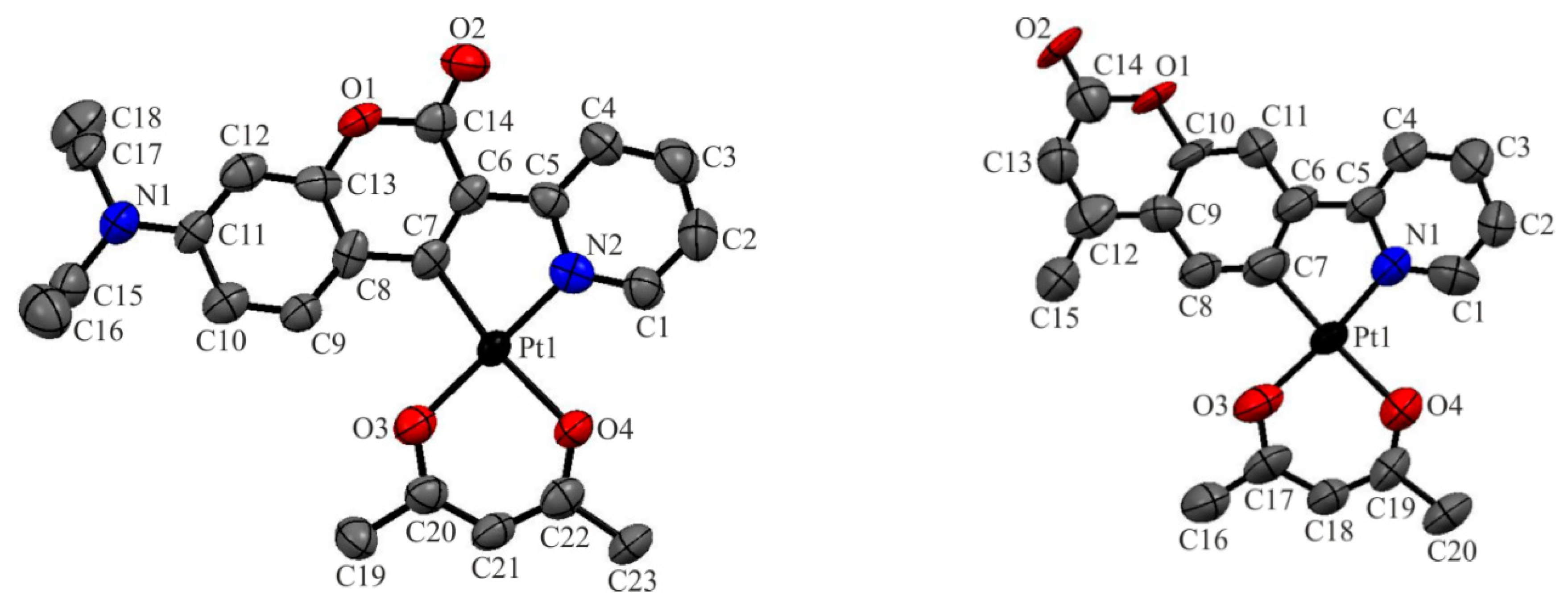
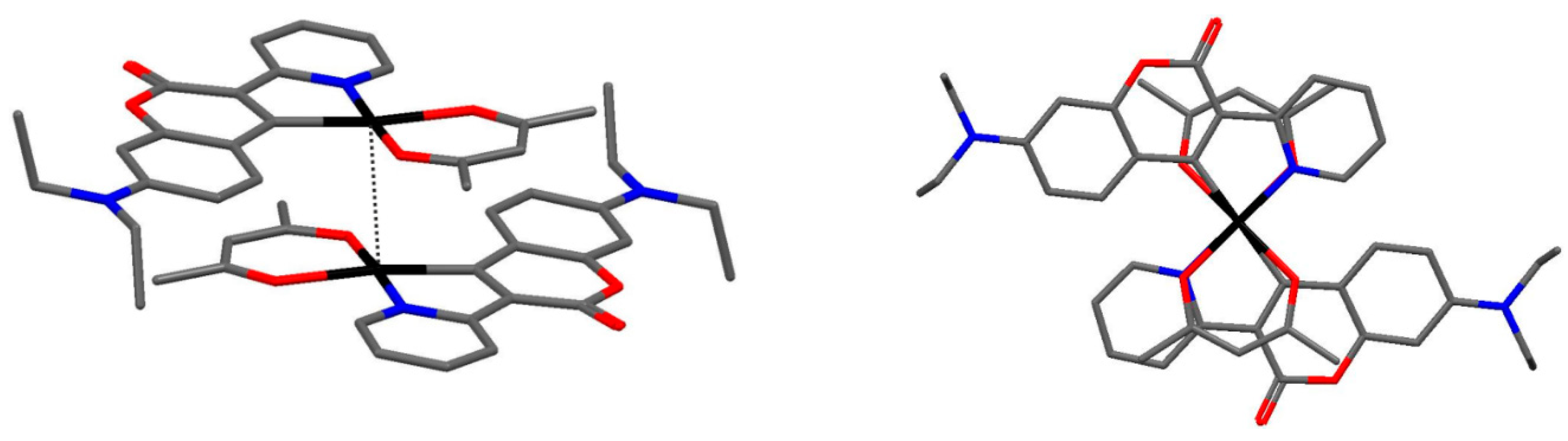
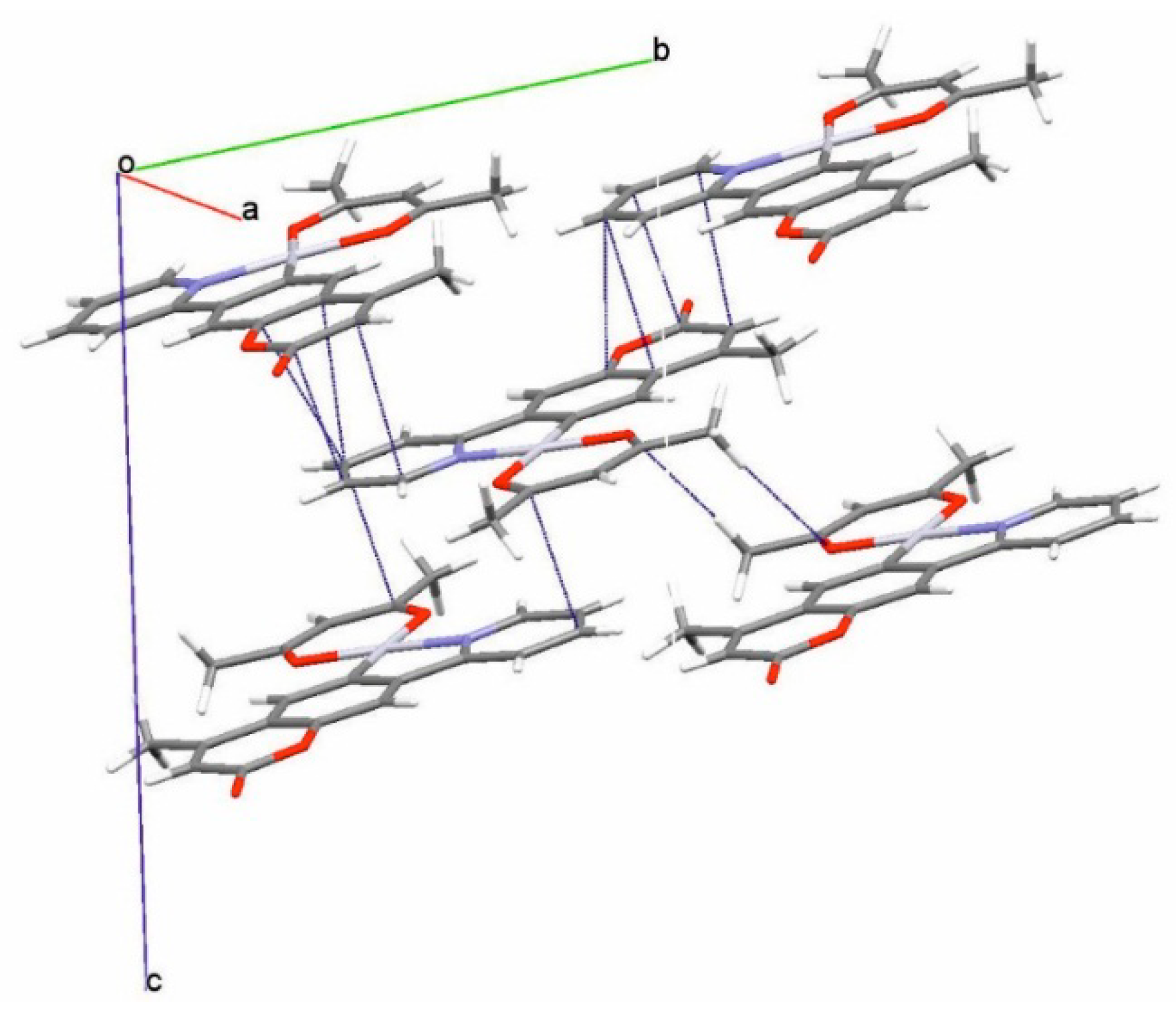
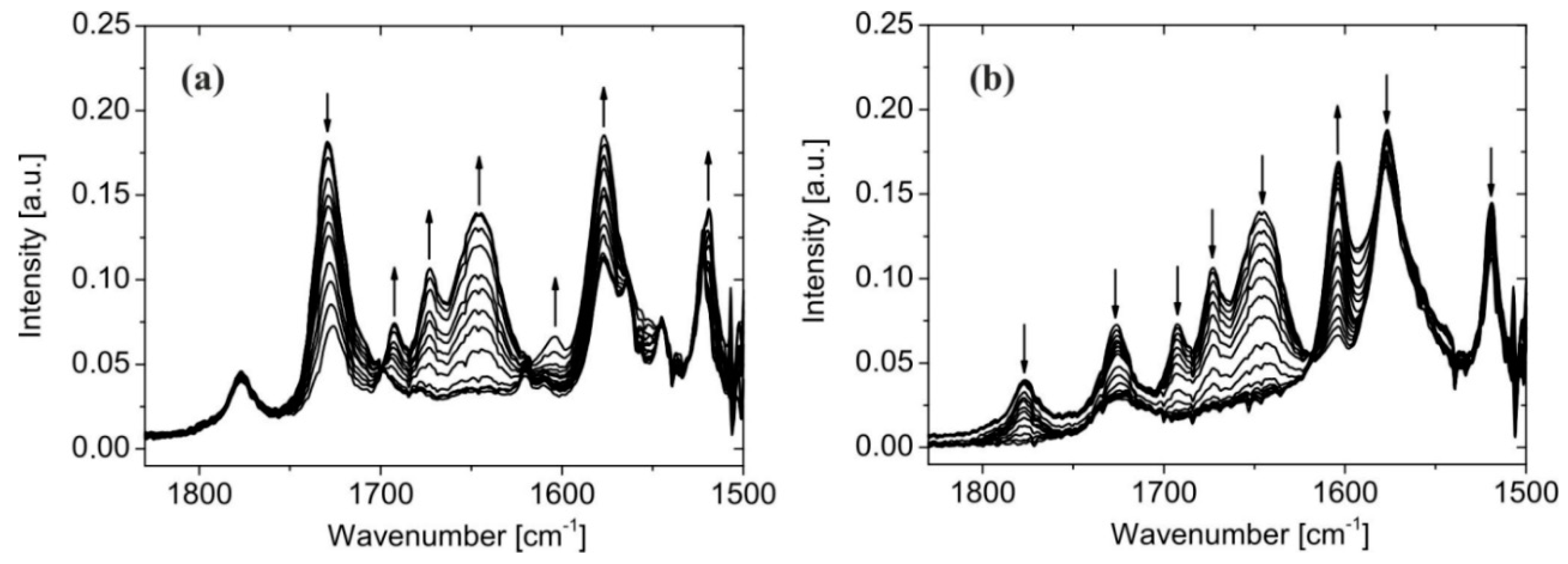
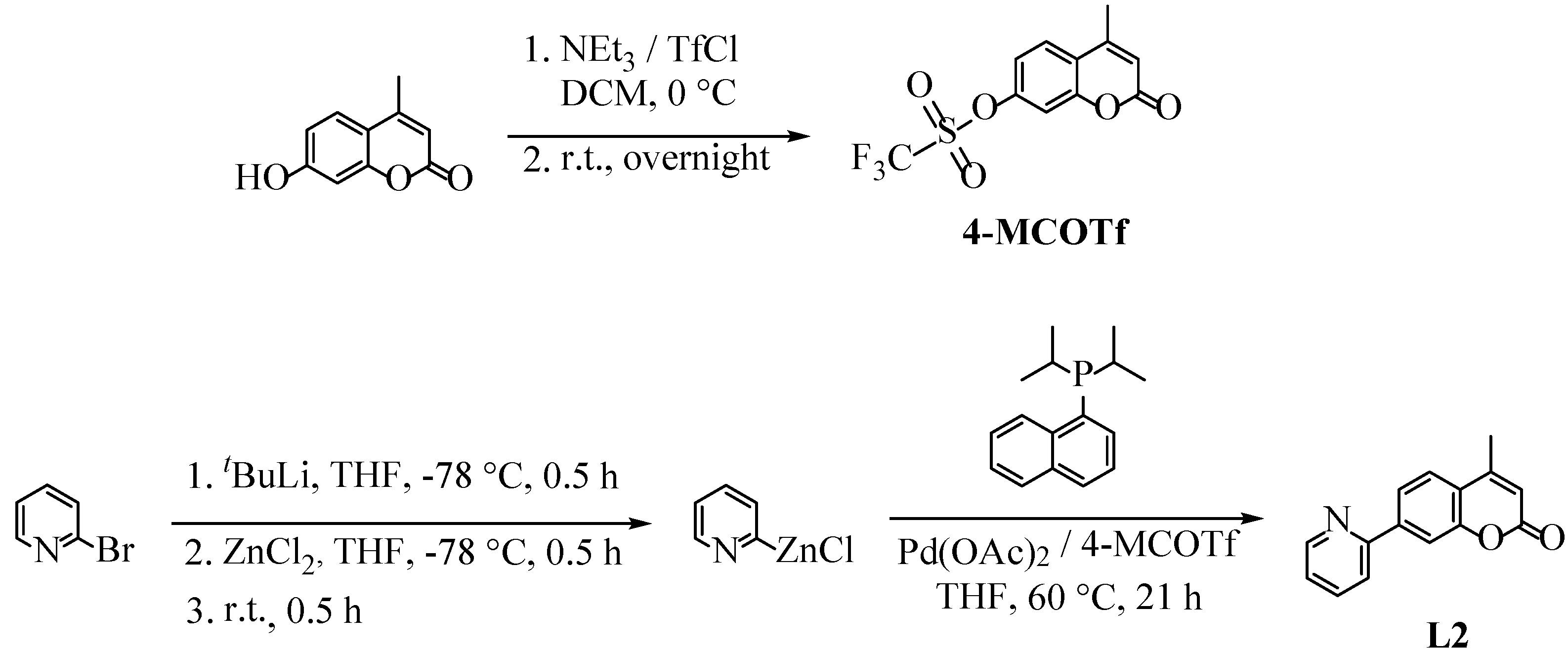
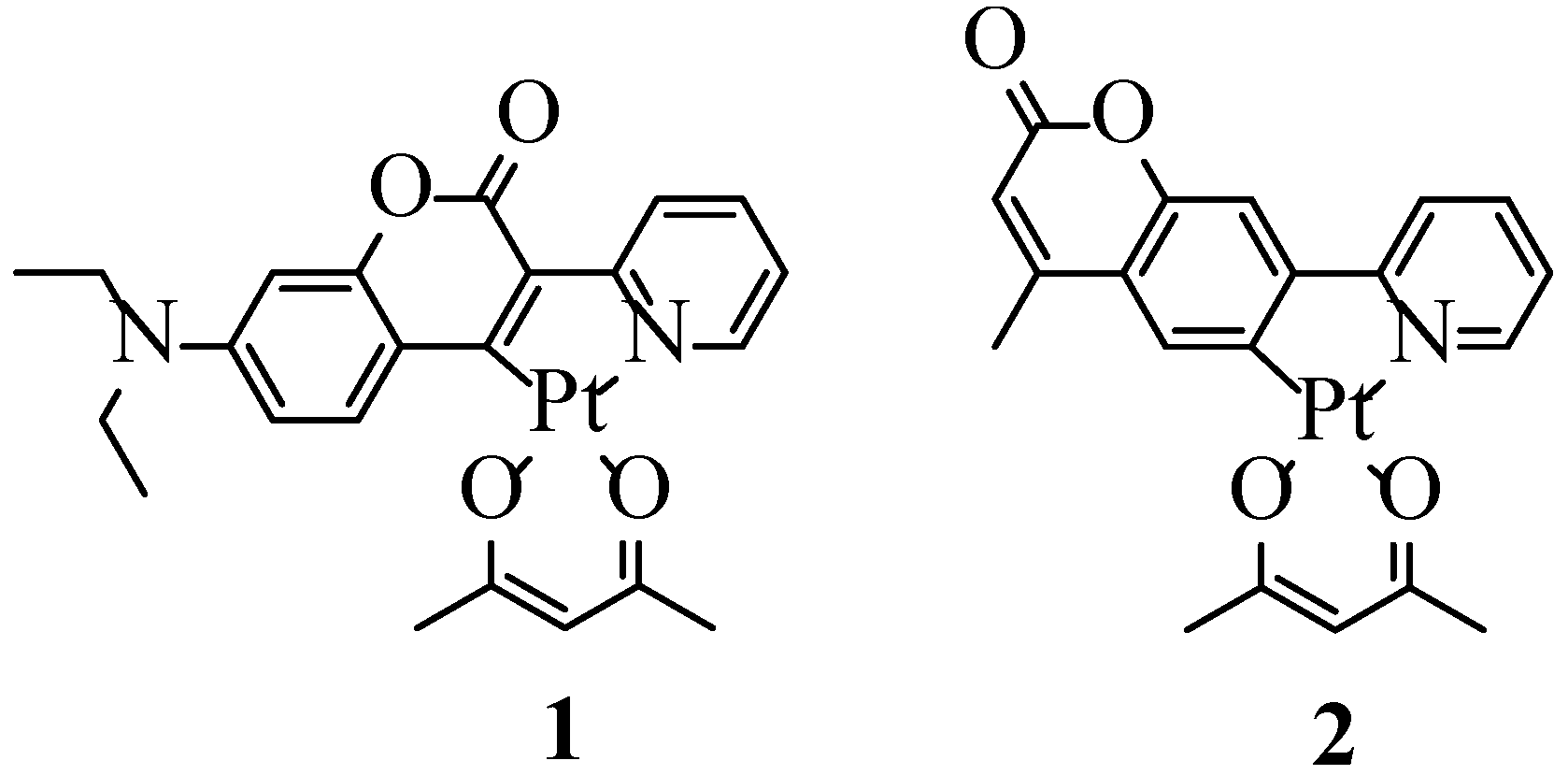
2.1. Synthesis, NMR Spectroscopy, and Structural Characterization





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
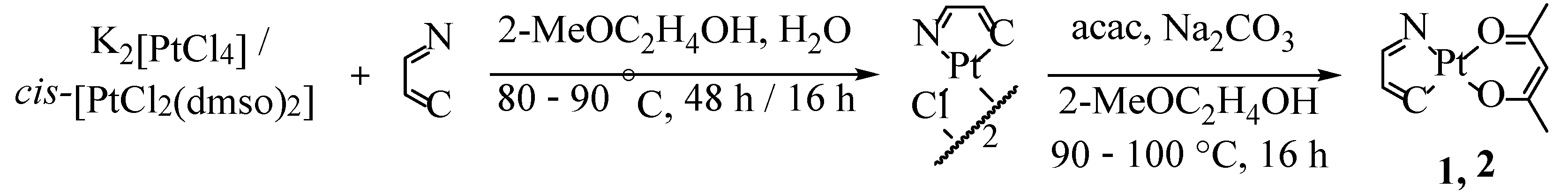{kind=link}
{kind=link}
| Bond parameter | 1 | Bond parameter | 2 |
|---|---|---|---|
| Pt(1)-N(2) | 1.974(5) | Pt(1)-N(1) | 1.965(11) |
| Pt(1)-C(7) | 1.978(6) | Pt(1)-C(7) | 1.983(9) |
| Pt(1)-O(3) | 2.013(4) | Pt(1)-O(3) | 2.010(9) |
| Pt(1)-O(4) | 2.072(4) | Pt(1)-O(4) | 2.089(8) |
| O(1)-C(13) | 1.379(7) | O(1)-C(13) | 1.371(13) |
| O(1)-C(14) | 1.382(7) | O(1)-C(14) | 1.395(11) |
| O(2)-C(14) | 1.221(7) | O(2)-C(14 | 1.241(14) |
| O(3)-C(20) | 1.292(7) | O(3)-C(17) | 1.284(13) |
| O(4)-C(22) | 1.279(7) | O(4)-C(19) | 1.283(14) |
| C(7)-Pt(1)-N(2) | 81.3(2) | C(7)-Pt(1)-N(1) | 82.4(4) |
| C(7)-Pt(1)-O(3) | 99.0(2) | C(7)-Pt(1)-O(3) | 91.2(4) |
| N(2)-Pt(1)-O(4) | 89.64(18) | N(1)-Pt(1)-O(4) | 93.5(3) |
| O(3)-Pt(1)-O(4) | 89.91(16) | O(3)-Pt(1)-O(4) | 92.9(3) |
| N(2)-Pt(1)-O(3) | 176.67(16) | N(1)-Pt(1)-O(3) | 173.6(3) |
| C(7)-Pt(1)-O(4) | 170.40(18) | C(7)-Pt(1)-O(4) | 175.9(4) |
| O(2)-C(14)-O(1) | 114.7(5) | O(2)-C(14)-O(1) | 116.3(10) |
| N(2)-C(5)-C(6)-C(7) | 0.3(7) | N(1)-C(5)-C(6)-C(7) | -0.6(14) |
| N(2)-Pt(1)-O(4)-C(22) | -178.3(4) | N(1)-Pt(1)-O(4)-C(19) | 178.0(9) |
| C(7)-Pt(1)-O(3)-C(20) | 176.8(4) | C(7)-Pt(1)-O(3)-C(17) | -179.3(10) |
| O(3)-Pt(1)-C(7)-C(8) | 4.9(5) | O(3)-Pt(1)-C(7)-C(8) | 1.0(12) |
| O(4)-Pt(1)-N(2)-C(5) | 173.0(4) | O(4)-Pt(1)-N(1)-C(5) | 179.2(8) |


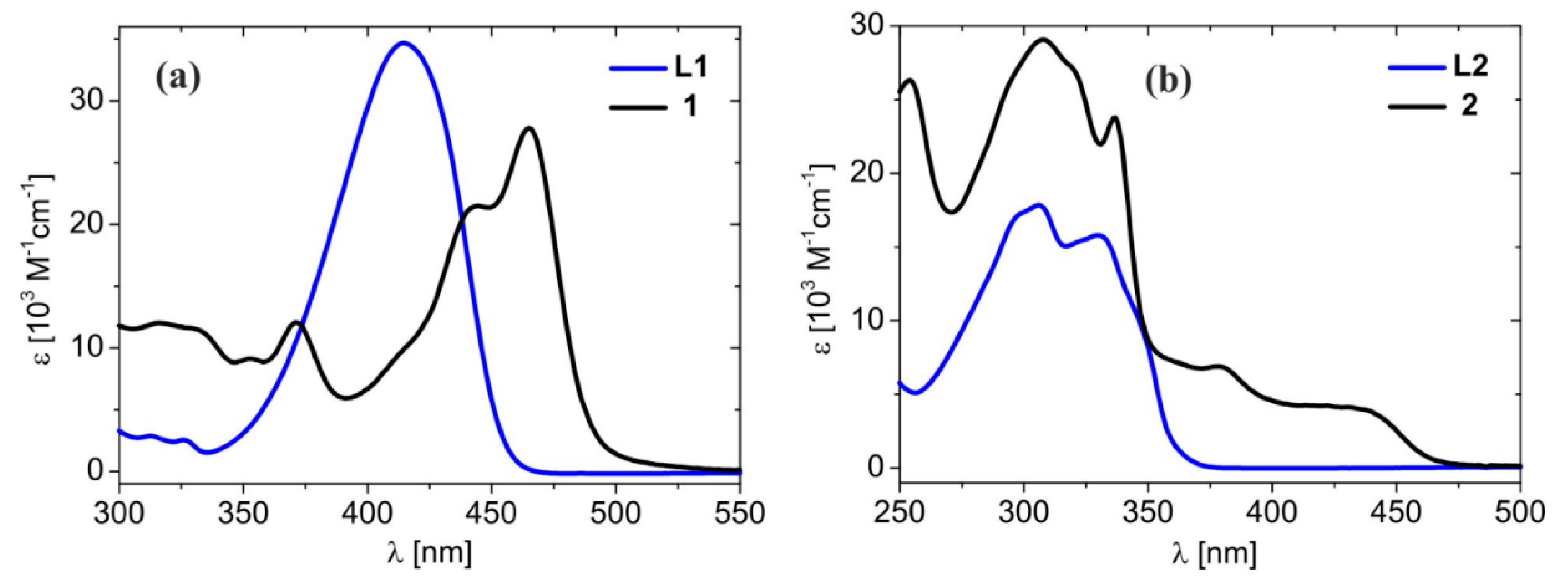
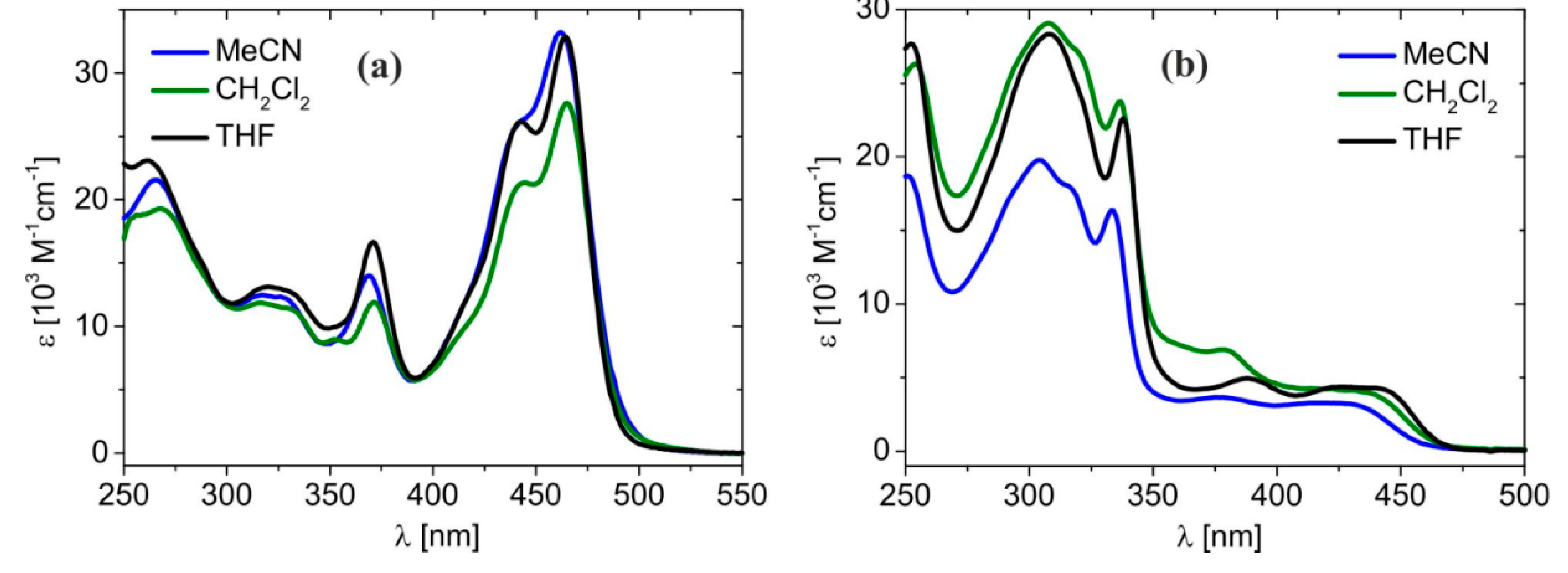
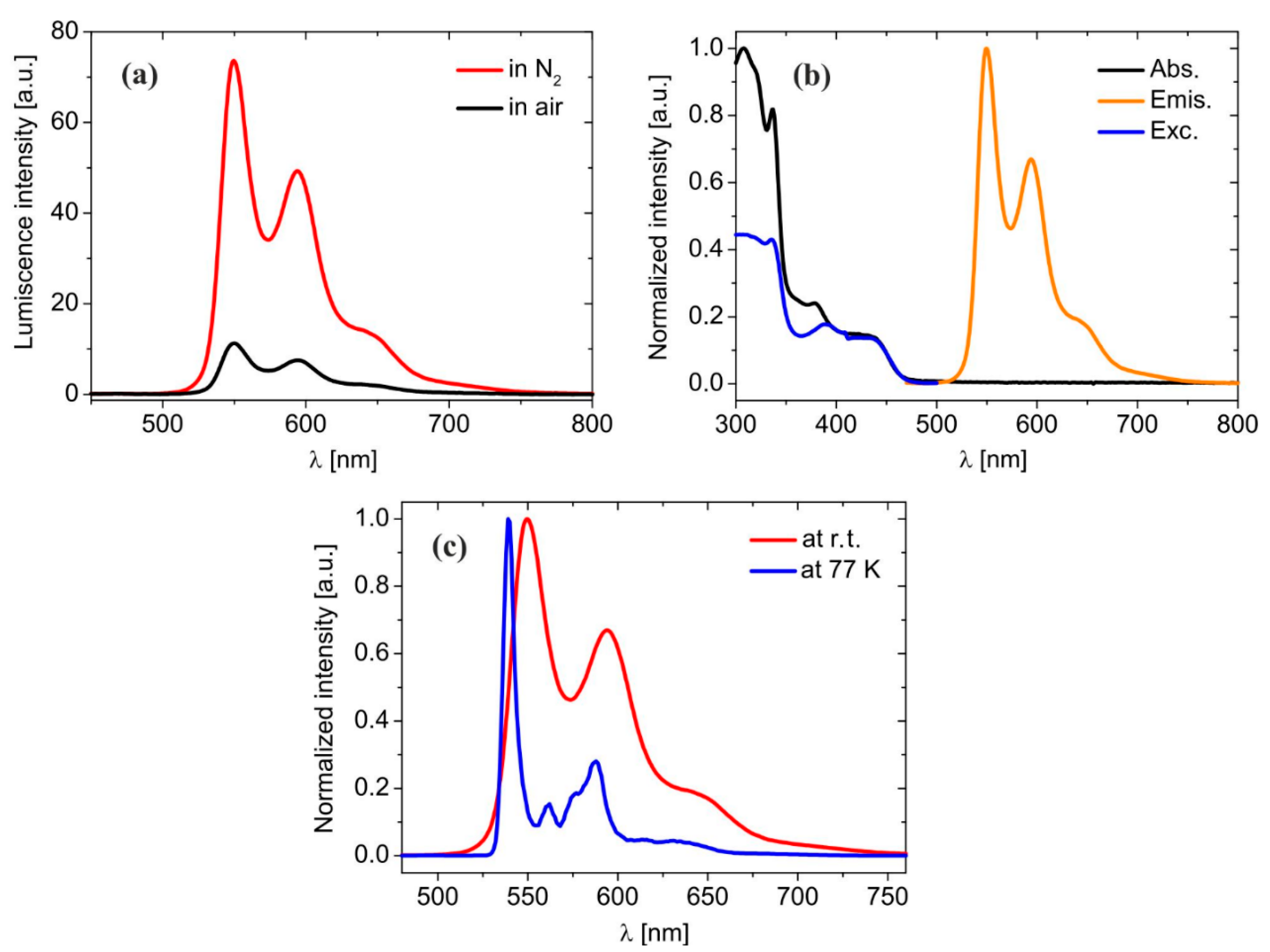
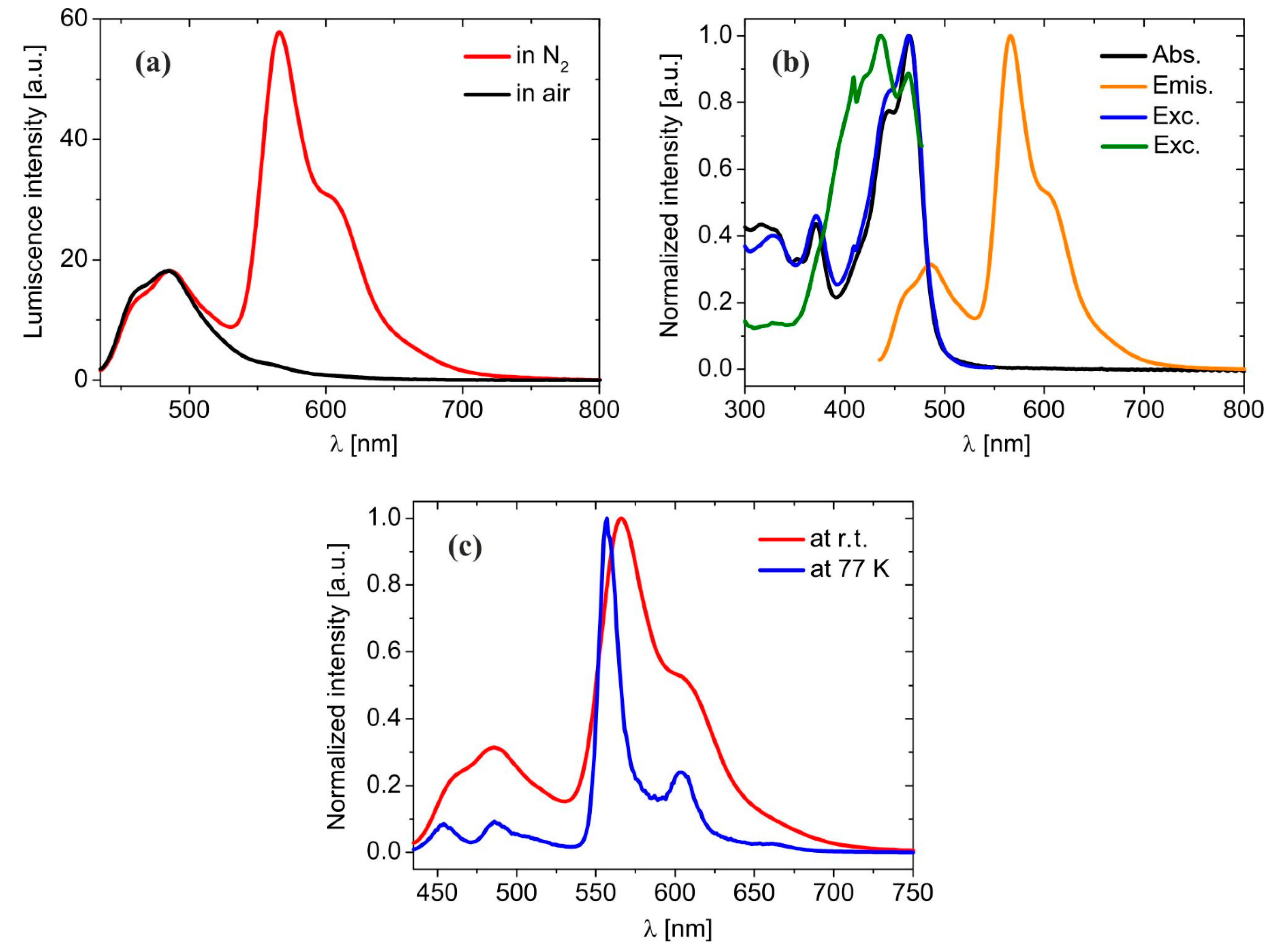
2.2. Absorption and Emission Properties

| Compound | Solvent | λ max [nm], (ε max [104 M−1cm−1]) |
|---|---|---|
| L1 | DCM | 273 (1.05), 414 (3.50) |
| L2 | DCM | 223 (2.05), 306 (1.78), 330 (1.58) |
| 1 | MeCN | 265 (2.16), 317 (1.25), 369 (1.40), 462 (3.32) |
| DCM | 268 (1.93), 316 (1.18), 371 (1.19), 465 (2.76) | |
| THF | 262 (2.31), 320 (1.31), 371 (1.67), 464 (3.29) | |
| 2 | MeCN | 251 (1.87), 304 (1.98), 333 (1.64), 377 (0.37), 416 (0.33), 426 (0.33) |
| DCM | 254 (2.63), 308 (2.91), 337 (2.38), 379 (0.69), 422 (0.42), 432 (0.41) | |
| THF | 252 (2.77), 308 (2.83), 338 (2.26), 388 (0.50), 427 (0.44), 439 (0.43) |





| Compound | λ max [nm], (ε max [104 M−1cm−1]) | λ em r.t. / 77 K [nm] | Φ F / P | τ r.t./77 K [μs] | |
|---|---|---|---|---|---|
| L1 | 414 (3.50) | 470 F | 0.83 [38] | 0.0026 | |
| L2 | 223 (2.05), 306 (1.78), 330 (1.58) | 376 F | 0.036 | - | |
| 1 | 268 (1.95), 316 (1.20), 371 (1.21), 465 (2.78) | 457 F, 486 F, 566/454F, 486F, 557454 F, 486 F, 557 | 0.064/0.210 | 0.0026F12.4/103.3 | |
| 2 | 254 (2.63), 308 (2.91), 337 (2.38), 379 (0.69), 422 (0.42) 432 (0.41) | 550/539 | -/0.213 | 1.6/15.3 | |


3. Experimental Section
3.1. Materials and General Methods
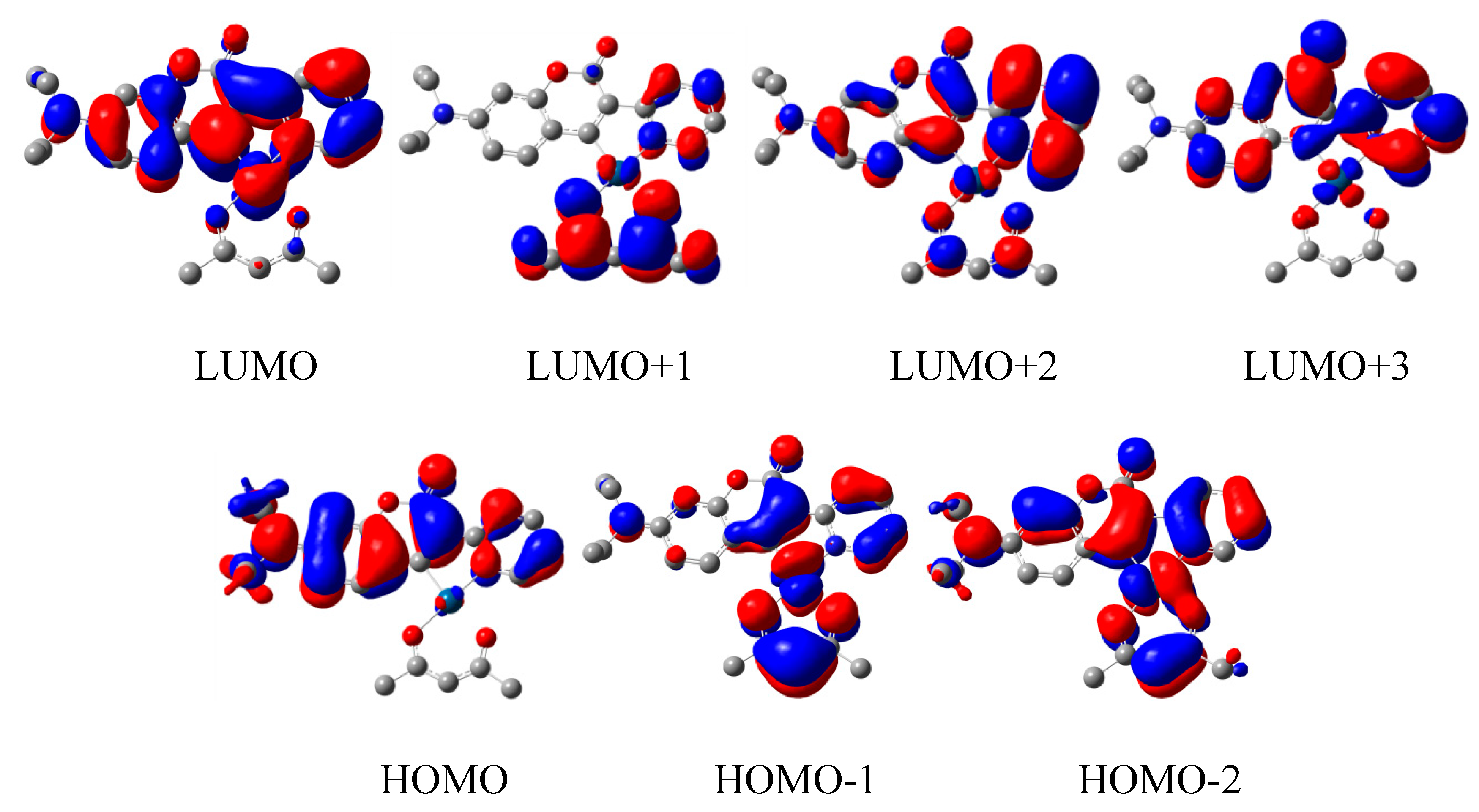
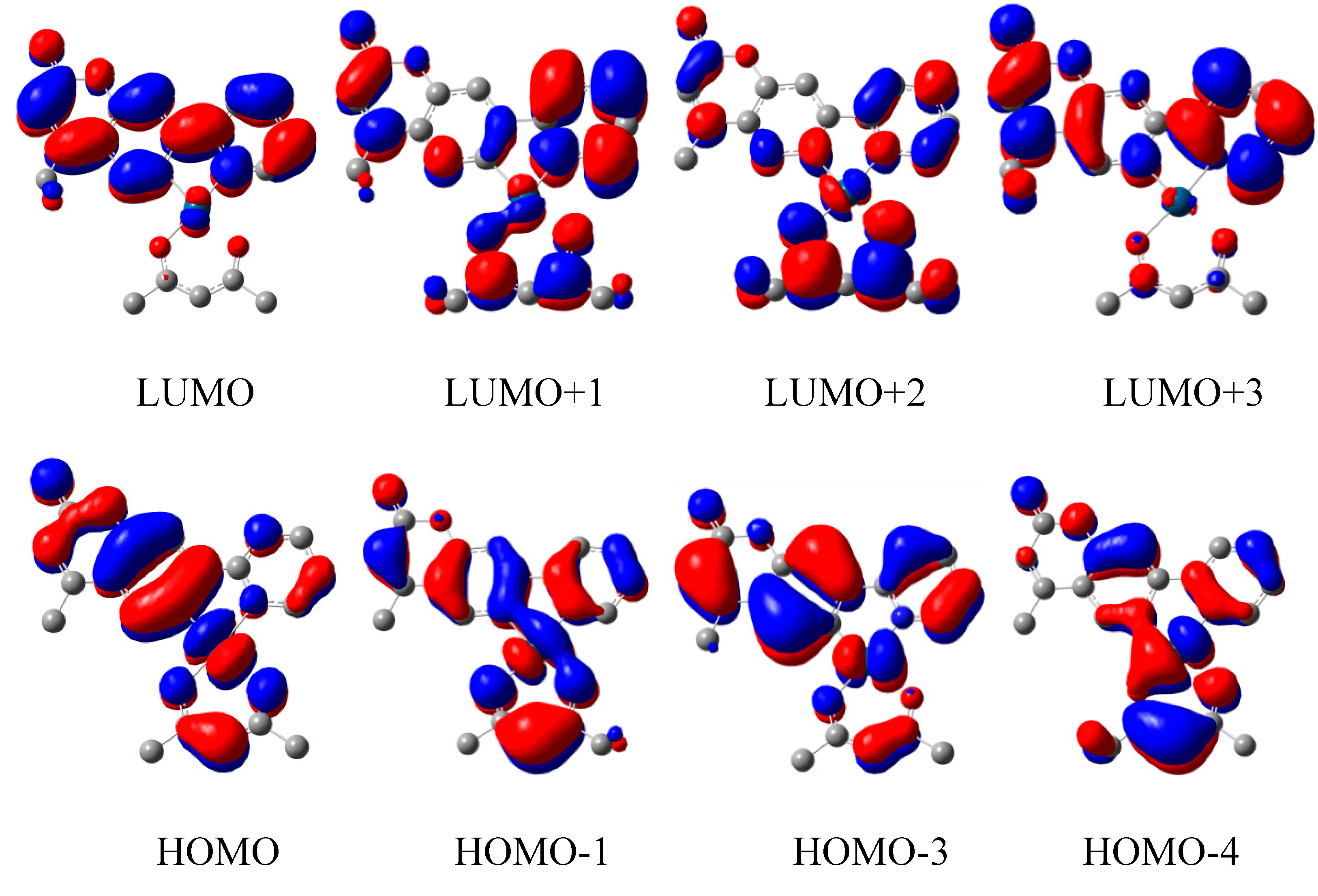
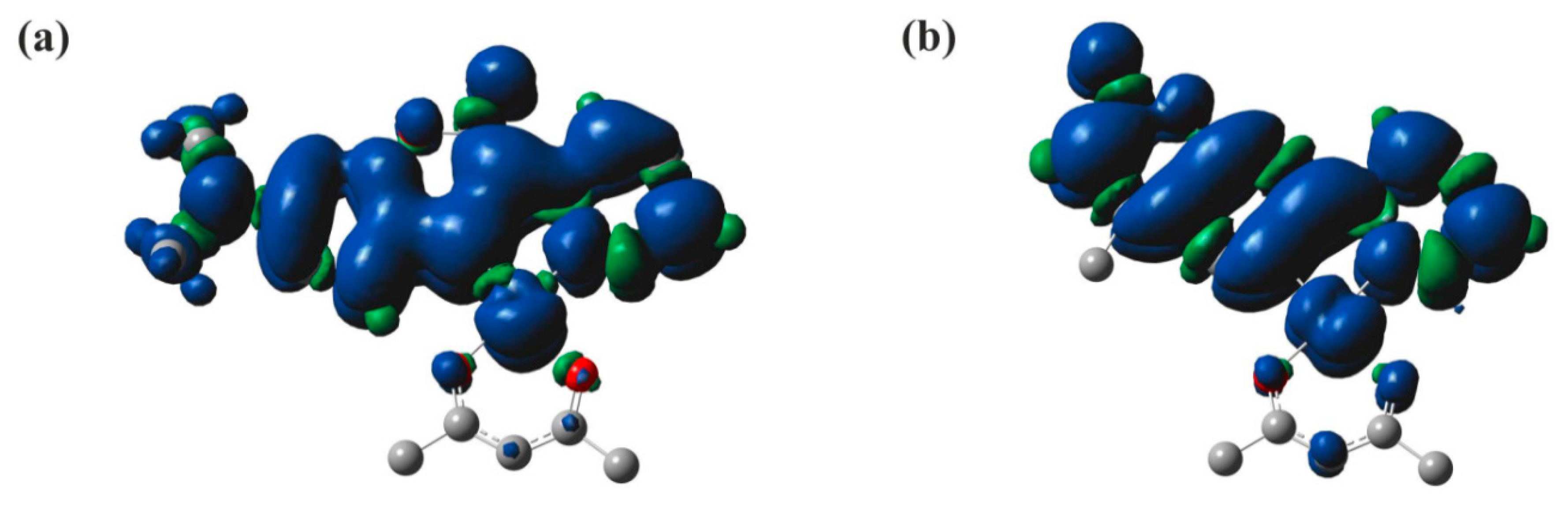
3.2. Quantum Chemical Calculations






Acknowledgements
Author Contributions
Appendix
References
- Brooks, W.J.; Babayan, V.; Lemansky, S.; Djurovich, P.I.; Tsyba, I.; Bau, R.; Thompson, M.E. Synthesis and Characterization of Phosphorescent Cyclometalated Platinum Complexes. Inorg. Chem. 2002, 41, 3055–3066. [Google Scholar] [PubMed]
- Djurovich, P.I.; Murphy, D.; Thompson, M.E.; Hernandez, B.; Gao, R.; Hunt, P.L.; Selke, M. Cyclometalated iridium and platinum complexes as singlet oxygen photosensitizers: Quantum yields, quenching rates and correlation with electronic structures. Dalton Trans. 2007, 3763–3770. [Google Scholar]
- Bossi, A.; Rausch, A.F.; Leitl, M.J.; Czerwieniec, R.; Whited, M.T.; Djurovich, P.I.; Yersin, H.; Thompson, M.E. Photophysical Properties of Cyclometalated Pt(II) Complexes: Counterintuitive Blue Shift in Emission with an Expanded Ligand π System. Inorg. Chem. 2013, 52, 12403–12415. [Google Scholar] [CrossRef] [PubMed]
- Ionkin, A.S.; Marshall, W.J.; Wang, Y. Syntheses, Structural Characterization, and First Electroluminescent Properties of Mono-cyclometalated Platinum(II) Complexes with Greater than Classical π−π Stacking and Pt−Pt Distances. Organometallics 2005, 24, 619–627. [Google Scholar] [CrossRef]
- He, Z.; Wong, W.-Y.; Yu, X.; Kwok, H.-S.; Lin, Z. Phosphorescent Platinum(II) Complexes Derived from Multifunctional Chromophores: Synthesis, Structures, Photophysics, and Electroluminescence. Inorg. Chem. 2006, 45, 10922–10937. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Niemeyer, F.; Williams, J.A.G.; Jiang, J.; Boucekkine, A.; Toupet, L.; Le Bozec, H.; Guerchais, V. Synthesis, Structure, and Photophysical Properties of Luminescent Platinum(II) Complexes Containing Cyclometalated 4-Styryl-Functionalized 2-Phenylpyridine Ligands. Inorg. Chem. 2006, 45, 8584–8596. [Google Scholar] [CrossRef]
- Chang, S.-Y.; Cheng, Y.-M.; Chi, Y.; Lin, Y.-C.; Jiang, C.-M.; Lee, G.-H.; Chou, P.-T. Emissive Pt(II) complexes bearing both cyclometalated ligand and 2-pyridyl hexafluoropropoxide ancillary chelate. Dalton Trans. 2008, 6901–6911. [Google Scholar] [CrossRef]
- Kozhevnikov, D.N.; Kozhevnikov, V.N.; Ustinova, M.M.; Santoro, A.; Bruce, D.W.; Koenig, B.; Czerwieniec, R.; Fischer, T.; Zabel, M.; Yersin, H. Synthesis of Cyclometallated Platinum Complexes with Substituted Thienylpyridines and Detailed Characterization of Their Luminescence Properties. Inorg. Chem. 2009, 48, 4179–4189. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Guo, H.; Ji, S.; Zhao, J. Long-Lived Room Temperature Deep-Red/Near-IR Emissive Intraligand Triplet Excited State (3IL) of Naphthalimide in Cyclometalated Platinum(II) Complexes and Its Application in Upconversion. Inorg. Chem. 2011, 50, 11446–11460. [Google Scholar] [CrossRef]
- Tronnier, A.; Risler, A.; Langer, N.; Wagenblast, G.; Münster, I.; Strassner, T. A Phosphorescent C^C* Cyclometalated Platinum(II) Dibenzothiophene NHC Complex. Organometallics 2012, 31, 7447–7452. [Google Scholar] [CrossRef]
- Uesugi, H.; Tsukuda, T.; Takao, K.; Tsubomura, T. Highly emissive platinum(II) complexes bearing carbene and cyclometalated ligands. Dalton Trans. 2013, 42, 7396–7403. [Google Scholar]
- Culham, S.; Lanoë, P.-H.; Whittle, V.L.; Durrant, M.C.; Williams, J.A.G.; Kozhevnikov, V.N. Highly Luminescent Dinuclear Platinum(II) Complexes Incorporating Bis-Cyclometallating Pyrazine-Based Ligands: A Versatile Approach to Efficient Red Phosphors. Inorg. Chem. 2013, 52, 10992–11003. [Google Scholar] [CrossRef] [PubMed]
- Moussa, J.; Cheminel, T.; Freeman, G.R.; Chamoreau, L.-M.; Williams, J.A.G.; Amouri, H. An unprecedented cyclometallated platinum(II) complex incorporating a phosphinine co-ligand: synthesis and photoluminescence behaviour. Dalton Trans. 2014, 43, 8162–8165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micksch, M.; Tenne, M.; Strassner, T. C^N-Cyclometalated Platinum(II) Complexes with Sterically Demanding 1,2-Diarylimidazole Ligands. Organometallics 2014, 33, 3464–3473. [Google Scholar] [CrossRef]
- Yu, J.; He, K.; Li, Y.; Tan, H.; Zhu, M.; Wang, Y.; Liu, Y.; Zhu, W.; Wu, H. A novel near-infrared-emitting cyclometalated platinum(II) complex with donor–acceptor–acceptor chromophores. Dyes Pigments 2014, 107, 146–152. [Google Scholar] [CrossRef]
- Liu, C.; Song, X.; Rao, X.; Xing, Y.; Wang, Z.; Zhao, J.; Qiu, J. Novel triphenylamine-based cyclometalated platinum(II) complexes for efficient luminescent oxygen sensing. Dyes Pigm. 2014, 101, 85–92. [Google Scholar] [CrossRef]
- Wu, W.; Wu, W.; Ji, S.; Guo, H.; Song, P.; Han, K.; Chi, L.; Shao, J.; Zhao, J. Tuning the emission properties of cyclometalated platinum(II) complexes by intramolecular electron-sink/arylethynylated ligands and its application for enhanced luminescent oxygen sensing. J. Mater. Chem. 2010, 20, 9775–9786. [Google Scholar] [CrossRef]
- Lai, S.-W.; Che, C.-M. Luminescent Cyclometalated Diimine Platinum(II) Complexes: Photophysical Studies and Applications. Top. Curr. Chem. 2004, 241, 27–63. [Google Scholar]
- Williams, J.A.G. Photochemistry and Photophysics of Coordination Compounds: Platinum. Top. Curr. Chem. 2007, 281, 205–268. [Google Scholar]
- Thompson, M.E.; Djurovich, P.I.; Barlow, S.; Marder, S.R. Organometallic Complexes for optoelectronic applications. In Comprehensive Organometallic Chemistry; O’Hare, D., Ed.; Elsevier: Oxford, UK, 2007; Volume 12, pp. 101–194. [Google Scholar]
- Chi, Y.; Chou, P.-T. Transition-metal phosphors with cyclometalating ligands: Fundamentals and applications. Chem. Soc. Rev. 2010, 39, 638–655. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.-Y.; Ho, C.-L. Heavy metal organometallic electrophosphors derived from multi-component chromophores. Coord. Chem. Rev. 2009, 253, 1709–1758. [Google Scholar] [CrossRef]
- Yersin, H.; Rausch, A.F.; Czerwieniec, R.; Hofbeck, T.; Fischer, T. The triplet state of organo-transition metal compounds. Triplet harvesting and singlet harvesting for efficient OLEDs. Coord. Chem. Rev. 2011, 255, 2622–2652. [Google Scholar] [CrossRef]
- Bronner, C.; Baudron, S.A.; Hosseini, M.W.; Strassert, C.A.; Guenet, A.; de Cola, L. Dipyrrin based luminescent cyclometallated palladium and platinum complexes. Dalton Trans. 2010, 39, 180–184. [Google Scholar] [CrossRef]
- Bronner, C.; Veiga, M.; Guenet, A.; de Cola, L.; Hosseini, M.W.; Strassert, C.A.; Baudron, S.A. Excited State Properties and Energy Transfer within Dipyrrin-Based Binuclear Iridium/Platinum Dyads: The Effect of ortho-Methylation on the Spacer. Chem. Eur. J. 2012, 18, 4041–4050. [Google Scholar] [CrossRef] [PubMed]
- Naziruddin, A.R.; Galstyan, A.; Iordache, A.; Daniliuc, C.G.; Strassert, C.A.; de Cola, L. Bidentate NHC^pyrozolate ligands in luminescent platinum(II) complexes. Dalton Trans. 2015. [Google Scholar] [CrossRef]
- Zhou, G.-J.; Wang, Q.; Wong, W.-Y.; Ma, D.; Wang, L.; Lin, Z. A versatile color tuning strategy for iridium(III) and platinum(II) electrophosphors by shifting the charge-transfer states with an electron-deficient core. J. Mater. Chem. 2009, 19, 1872–1883. [Google Scholar] [CrossRef]
- Tam, A.Y.-Y.; Tsang, D.P.-K.; Chan, M.-Y.; Zhu, N.; Yam, V.W.-W. A luminescent cyclometalated platinum(II) complex and its green organic light emitting device with high device performance. Chem. Commun. 2011, 47, 3383–3385. [Google Scholar] [CrossRef]
- Wu, W.; Cheng, C.; Wu, W.; Guo, H.; Ji, S.; Song, P.; Han, K.; Zhao, J.; Zhang, X.; Wu, Y.; Du, G. Tuning the Emission Colour of Triphenylamine-Capped Cyclometallated Platinum(II) Complexes and Their Application in Luminescent Oxygen Sensing and Organic Light-Emitting Diodes. Eur. J. Inorg. Chem. 2010, 2010, 4683–4696. [Google Scholar] [CrossRef]
- Wu, W.; Wu, W.; Ji, S.; Guo, H.; Zhao, J. Accessing the long-lived emissive 3IL triplet excited states of coumarin fluorophores by direct cyclometallation and its application for oxygen sensing and upconversion. Dalton Trans. 2011, 40, 5953–5963. [Google Scholar] [CrossRef] [PubMed]
- Pevny, F.; Zabel, M.; Winter, R.F.; Rausch, A.F.; Yersin, H.; Tuczek, F.; Záliš, S. Improvement of (bipy)Pt(XR)2 (X = O, S) type photosensitizers by covalent dye attachment. Chem. Commun. 2011, 6302–6304. [Google Scholar] [CrossRef]
- Wu, W.; Ji, S.; Wu, W.; Shao, J.; Guo, H.; James, T.D.; Zhao, J. Ruthenium(II)–Polyimine–Coumarin Light-Harvesting Molecular Arrays: Design Rationale and Application for Triplet–Triplet-Annihilation-Based Upconversion. Chem. Eur. J. 2012, 18, 4953–4964. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Qin, H.; Chen, H.; Sun, H.; Zhao, J. Phenylacetylide ligand mediated tuning of visible-light absorption, room temperature phosphorescence lifetime and triplet–triplet annihilation based up-conversion of a diimine Pt(II) bisacetylide complex. Dyes Pigments 2013, 99, 908–915. [Google Scholar] [CrossRef]
- Sun, H.; Guo, H.; Wu, W.; Liu, X.; Zhao, J. Coumarin phosphorescence observed with N^NPt(II) bisacetylide complex and its applications for luminescent oxygen sensing and triplet-triplet-annihilation based upconversion. Dalton Trans. 2011, 40, 7834–7841. [Google Scholar] [CrossRef] [PubMed]
- Ulbricht, C.; Rehmann, N.; Holder, E.; Hertel, D.; Meerholz, K.; Schubert, U.S. Synthesis and Characterization of Oxetane-Functionalized Phosphorescent Ir(III)-Complexes. Macromol. Chem. Phys. 2009, 210, 531–541. [Google Scholar] [CrossRef]
- Geist, F.; Jackel, A.; Winter, R.F. Dual ligand-based fluorescence and phosphorescence emission at room temperature from platinum thioxanthonyl complexes. Dalton Trans. 2015, 44, 3974–3987. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yang, S.; Zhao, Y.; Zhang, H.; Fan, D.; Han, X.; Liu, Z. Synthesis, crystal structure and photoluminescent property of an iridium complex with coumarin derivative ligand. Inorg. Chim. Acta 2011, 379, 171–174. [Google Scholar] [CrossRef]
- Yu, T.; Yang, S.; Zhao, Y.; Zhang, H.; Han, X.; Fan, D.; Qiu, Y.; Chen, L. Synthesis, crystal structures and fluorescence properties of 3-(2-pyridyl)coumarin derivatives. J. Photochem. Photobiol. A 2010, 214, 92–99. [Google Scholar] [CrossRef]
- Du, L.; Li, M.; Zheng, S.; Wang, B. Rational design of a fluorescent hydrogen peroxide probe based on the umbelliferone fluorophore. Tetrahedron Lett. 2008, 49, 3045–3048. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Manabe, K. Negishi coupling strategy of a repetitive two-step method for oligoarene synthesis. Tetrahedron Lett. 2006, 47, 5927–5931. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Buchwald, S.L. A Highly Active Catalyst for the Room-Temperature Amination and Suzuki Coupling of Aryl Chlorides. Angew. Chem. Int. Ed. Engl. 1999, 38, 2413–2416. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, B.N.; Howe, D.V.; Keating, T.; Johnson, B.F.G.; Lewis, J. Reactivity of co-ordinated ligands. Part XV. Formation of complexes containing Group V donor atoms and metal-carbon σ-bonds. Dalton Trans. 1973, 404–410. [Google Scholar] [CrossRef]
- Godbert, N.; Pugliese, T.; Aiello, I.; Bellusci, A.; Crispini, A.; Ghedini, M. Efficient, Ultrafast, Microwave-Assisted Syntheses of Cycloplatinated Complexes. Eur. J. Inorg. Chem. 2007, 2007, 5105–5111. [Google Scholar] [CrossRef]
- Mdleleni, M.M.; Bridgewater, J.S.; Watts, R.J.; Ford, P.C. Synthesis, Structure, and Spectroscopic Properties of Ortho-Metalated Platinum(II) Complexes. Inorg. Chem. 1995, 34, 2334–2342. [Google Scholar] [CrossRef]
- Ghedini, M.; Pucci, D.; Crispini, A.; Barberio, G. Oxidative Addition to Cyclometalated Azobenzene Platinum(II) Complexes: A Route to Octahedral Liquid Crystalline Materials. Organometallics 1999, 18, 2116–2124. [Google Scholar] [CrossRef]
- Li, K.; Cheng, G.; Ma, C.; Guan, X.; Kwok, W.-M.; Chen, Y.; Lu, W.; Che, C.-M. Light-emitting platinum(II) complexes supported by tetradentate dianionic bis(N-heterocyclic carbene) ligands: towards robust blue electrophosphors. Chem. Sci. 2013, 4, 2630–2644. [Google Scholar] [CrossRef]
- Wong, W.-W.; He, Z.; So, S.-K.; Tong, K.-L.; Lin, Z. A Multifunctional Platinum-Based Triplet Emitter for OLED Applications. Organometallics 2005, 24, 4079–4082. [Google Scholar] [CrossRef]
- Lai, S.-W.; Chan, M.C.-W.; Cheung, T.-C.; Peng, S.-M.; Che, C.-M. Probing d8−d8 Interactions in Luminescent Mono- and Binuclear Cyclometalated Platinum(II) Complexes of 6-Phenyl-2,2‘-bipyridines. Inorg. Chem. 1999, 38, 4046–4055. [Google Scholar] [CrossRef]
- Lu, W.; Chan, M.C.W.; Zhu, N.; Che, C.-M.; Li, C.; Hui, Z. Structural and Spectroscopic Studies on Pt···Pt and π−π Interactions in Luminescent Multinuclear Cyclometalated Platinum(II) Homologues Tethered by Oligophosphine Auxiliaries. J. Am. Chem. Soc. 2004, 126, 7639–7651. [Google Scholar] [CrossRef]
- Kui, S.C.F.; Chui, S.S.-Y.; Che, C.-M.; Zhu, N. Structures, Photoluminescence, and Reversible Vapoluminescence Properties of Neutral Platinum(II) Complexes Containing Extended π-Conjugated Cyclometalated Ligands. J. Am. Chem. Soc. 2006, 128, 8297–8309. [Google Scholar] [CrossRef] [PubMed]
- Kui, S.C.F.; Sham, I.H.T.; Cheung, C.C.C.; Ma, C.-W.; Yan, B.; Zhu, N.; Che, C.-M.; Fu, W.-F. Platinum(II) Complexes with π-Conjugated, Naphthyl-Substituted, Cyclometalated Ligands (RC^N^N): Structures and Photo- and Electroluminescence. Chem. Eur. J. 2007, 7, 417–435. [Google Scholar] [CrossRef]
- Miller, J.S.; Epstein, A.J. One-Dimensional Inorganic Complexes. In Progress in Inorganic Chemistry; John Wiley & Sons, Inc.: Weinheim, Germany, 1976; pp. 1–151. [Google Scholar]
- Sun, Y.; Ye, K.; Zhang, H.; Zhang, J.; Zhao, L.; Li, B.; Yang, G.; Yang, B.; Wang, Y.; Lai, S.-W.; Che, C.-M. Luminescent One-Dimensional Nanoscale Materials with PtII⋅⋅⋅PtII Interactions. Angew. Chem. Int. Ed. 2006, 45, 5610–5613. [Google Scholar]
- Rodionova, O.A.; Puzyk, M.V.; Balashev, K.P. Effect of cyclopalladation on the spectroscopic properties of a coumarin dye. Opt. Spectrosc. 2008, 105, 62–66. [Google Scholar] [CrossRef]
- Kaim, W.; Ernst, S.; Kohlmann, S. Farbige Komplexe: das Charge-Transfer Phänomen. Chem. Uns. Zeit 1987, 21, 50–58. [Google Scholar] [CrossRef]
- Krejcik, M.; Danek, M.; Hartl, F. Simple construction of an infrared optically transparent thin-layer cell: Applications to the redox reactions of ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-di-t-butyl-catecholate)−. J. Electroanal. Chem. 1991, 317, 179–187. [Google Scholar] [CrossRef]
- Rachford, A.A.; Goeb, S.; Castellano, F.N. Accessing the Triplet Excited State in Perylenediimides. J. Am. Chem. Soc. 2008, 130, 2766–2767. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-Y.; Liu, S.; Köse, M.E.; Schanze, K.S. Photophysics of Platinum-Acetylide Substituted Hexa-peri-hexabenzacoronenes. Inorg. Chem. 2006, 45, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, D.; Draper, S.M.; Yi, X.; Wu, W.; Zhao, J. Visible light-harvesting trans bis(alkylphosphine) platinum(II)-alkynyl complexes showing long-lived triplet excited states as triplet photosensitizers for triplet-triplet annihilation upconversion. Dalton Trans. 2013, 42, 10694–10706. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Muro-Small, M.L.; Ji, S.; Zhao, J.; Castellano, F.N. Naphthalimide Phosphorescence Finally Exposed in a Platinum(II) Diimine Complex. Inorg. Chem. 2010, 49, 6802–6804. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhao, J.; Sun, J.; Huang, L.; Yi, X. Red-light excitable fluorescent platinum(II) bis(aryleneethynylene) bis(trialkylphosphine) complexes showing long-lived triplet excited states as triplet photosensitizers for triplet-triplet annihilation upconversion. J. Mater. Chem. C 2013, 1, 705–716. [Google Scholar] [CrossRef]
- Nguyen, M.-H.; Yip, J.H.K. Platinum-Conjugated Homo- and Heterobichromophoric Complexes of Tetracene and Pentacene. Organometallics 2012, 31, 7522–7531. [Google Scholar]
- Nguyen, M.-H.; Nguyen, V.H.; Yip, J.H.K. Sequence-Specific Synthesis of Platinum-Conjugated Trichromophoric Energy Cascades of Anthracene, Tetracene, and Pentacene and Fluorescent “Black Chromophores”. Organometallics 2013, 32, 7283–7291. [Google Scholar] [CrossRef]
- Tanaka, Y.; Wong, K.M.-C.; Yam, V.W.-W. Platinum-Based Phosphorescent Double-Decker Tweezers: A Strategy for Extended Heterologous Metal–Metal Interactions. Angew. Chem, Int. Ed. 2013, 52, 14117–14120. [Google Scholar] [CrossRef]
- Chan, A.K.-W.; Lam, E.S.-H.; Tam, A.Y.-Y.; Tsang, D.P.-K.; Lam, W.H.; Chan, M.-Y.; Wong, W.-T.; Yam, V.W.-W. Synthesis and Characterization of Luminescent Cyclometalated Platinum(II) Complexes of 1,3-Bis-Hetero-Azolylbenzenes with Tunable Color for Applications in Organic Light-Emitting Devices through Extension of π Conjugation by Variation of the Heteroatom. Chem. Eur. J. 2013, 19, 13910–13924. [Google Scholar] [CrossRef]
- Leung, S.Y.-L.; Lam, E.S.-H.; Lam, W.H.; Wong, K.M.-C.; Wong, W.-T.; Yam, V.W.-W. Luminescent Cyclometalated Alkynylplatinum(II) Complexes with a Tridentate Pyridine-Based N-Heterocyclic Carbene Ligand: Synthesis, Characterization, Electrochemistry, Photophysics, and Computational Studies. Chem. Eur. J. 2013, 19, 10360–10369. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.A.; Bhargava, S.K.; Cheng, E.C.-C.; Lam, W.H.; Lee, T.K.-M.; Privér, S.H.; Wagler, J.; Willis, A.C.; Yam, V.W.-W. Unprecedented Near-Infrared (NIR) Emission in Diplatinum(III) (d7−d7) Complexes at Room Temperature. J. Am. Chem. Soc. 2010, 132, 7094–7103. [Google Scholar] [CrossRef]
- Pomestchenko, I.E.; Luman, C.R.; Hissler, M.; Ziessel, R.; Castellano, F.N. Room Temperature Phosphorescence from a Platinum(II) Diimine Bis(pyrenylacetylide) Complex. Inorg. Chem. 2003, 42, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Kozhevnikov, D.N.; Kozhevnikov, V.N.; Shafikov, M.Z.; Prokhorov, A.M.; Bruce, D.W.; Gareth Williams, J.A. Phosphorescence vs. Fluorescence in Cyclometalated Platinum(II) and Iridium(III) Complexes of (Oligo)thienylpyridines. Inorg. Chem. 2011, 50, 3804–3815. [Google Scholar]
- Liu, Y.; Guo, H.; Zhao, J. Ratiometric luminescent molecular oxygen sensors based on uni-luminophores of C^N Pt(II)(acac) complexes that show intense visible-light absorption and balanced fluorescence/phosphorescence dual emission. Chem. Commun. 2011, 47, 11471–11473. [Google Scholar] [CrossRef]
- Chia, Y.Y.; Tay, M.G. An insight into fluorescent transition metal complexes. Dalton Trans. 2014, 43, 13159–13168. [Google Scholar] [CrossRef] [PubMed]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Compounds of general interest. Dimethylsulfoxide complexes of Pt(II): K[PtCl3(Me2SO], cis-[PtCl2L(Me2SO)] (L = Me2SO, MeCN), [PtCl(µ-Cl)(Me2SO)]2, and [Pt(Me2SO)4](CF3SO3)2. Inorg. Synth. 2002, 33, 192. [Google Scholar]
- Sheldrick, G.M. SHELX-97; Program for Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Frisch, M.J.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.01; Gaussian Inc.: Wallingford, CT, 2009. [Google Scholar]
- Gunnarsson, O.; Lundqvist, B.I. Exchange and correlation in atoms, molecules, and solids by the spin-density-functional formalism. Phys. Rev. B 1976, 13, 4274. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 1989, 90, 1730–1734. [Google Scholar]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100, 7535–7532. [Google Scholar] [CrossRef]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Hariharan, P.H.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Enzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Cancés, E.; Menucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Scalamani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110–114124. [Google Scholar] [CrossRef] [PubMed]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.; Millam, J. GaussView; Version 3; Semichem Inc.: Shawnee Mission KS, 2009. [Google Scholar]
- Hu, R.-B.; Zhang, H.; Zhang, X.-Y.; Yang, S.-D. Palladium-catalyzed P(O)R2 directed C-H arylation to synthesize electron-rich polyaromatic monophosphorus ligands. Chem. Commun. 2014, 50, 2193–2195. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackel, A.; Linseis, M.; Häge, C.; Winter, R.F. Turning-On of Coumarin Phosphorescence in Acetylacetonato Platinum Complexes of Cyclometalated Pyridyl-Substituted Coumarins. Inorganics 2015, 3, 55-81. https://doi.org/10.3390/inorganics3020055
Jackel A, Linseis M, Häge C, Winter RF. Turning-On of Coumarin Phosphorescence in Acetylacetonato Platinum Complexes of Cyclometalated Pyridyl-Substituted Coumarins. Inorganics. 2015; 3(2):55-81. https://doi.org/10.3390/inorganics3020055
Chicago/Turabian StyleJackel, Andrej, Michael Linseis, Christian Häge, and Rainer F. Winter. 2015. "Turning-On of Coumarin Phosphorescence in Acetylacetonato Platinum Complexes of Cyclometalated Pyridyl-Substituted Coumarins" Inorganics 3, no. 2: 55-81. https://doi.org/10.3390/inorganics3020055
APA StyleJackel, A., Linseis, M., Häge, C., & Winter, R. F. (2015). Turning-On of Coumarin Phosphorescence in Acetylacetonato Platinum Complexes of Cyclometalated Pyridyl-Substituted Coumarins. Inorganics, 3(2), 55-81. https://doi.org/10.3390/inorganics3020055