Ketone Formation via Decarboxylation Reactions of Fatty Acids Using Solid Hydroxide/Oxide Catalysts

 and
and
Abstract
:1. Introduction
1.1. An Introduction to Layered Double Hydroxides
1.2. Layered Double Hydroxides as Catalysts and Catalyst Precursors
2. Results
2.1. Structure of Prepared Mixed Metal Oxide Materials
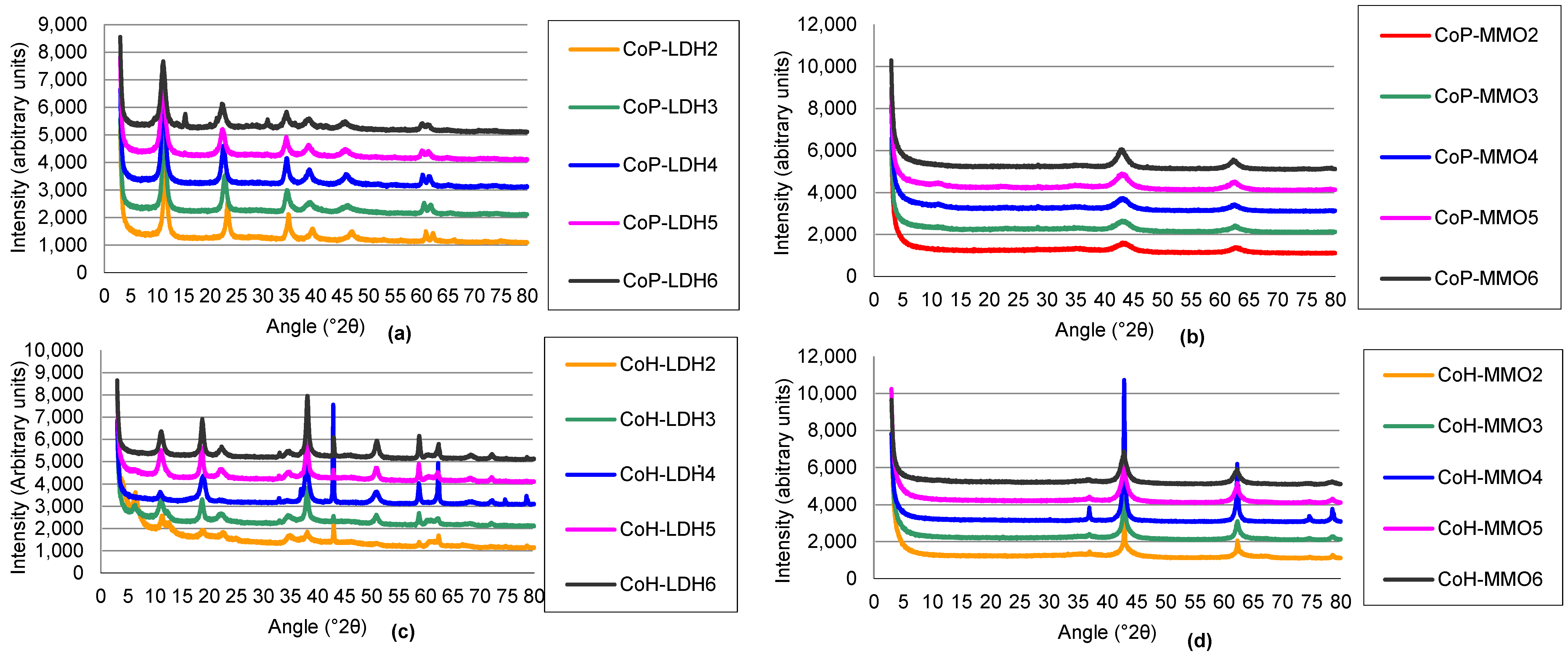
2.1.1. Structure and Basicity of Layered Double Hydroxides Prepared by Co-Precipitation
2.1.2. Structure and Basicity of Mixed Metal Oxides Prepared from Co-Precipitated Layered Double Hydroxides
2.1.3. Structure and Basicity of Layered Double Hydroxides Prepared by Co-Hydration
2.1.4. Mixed Metal Oxides Prepared from Co-Hydration Layered Double Hydroxides
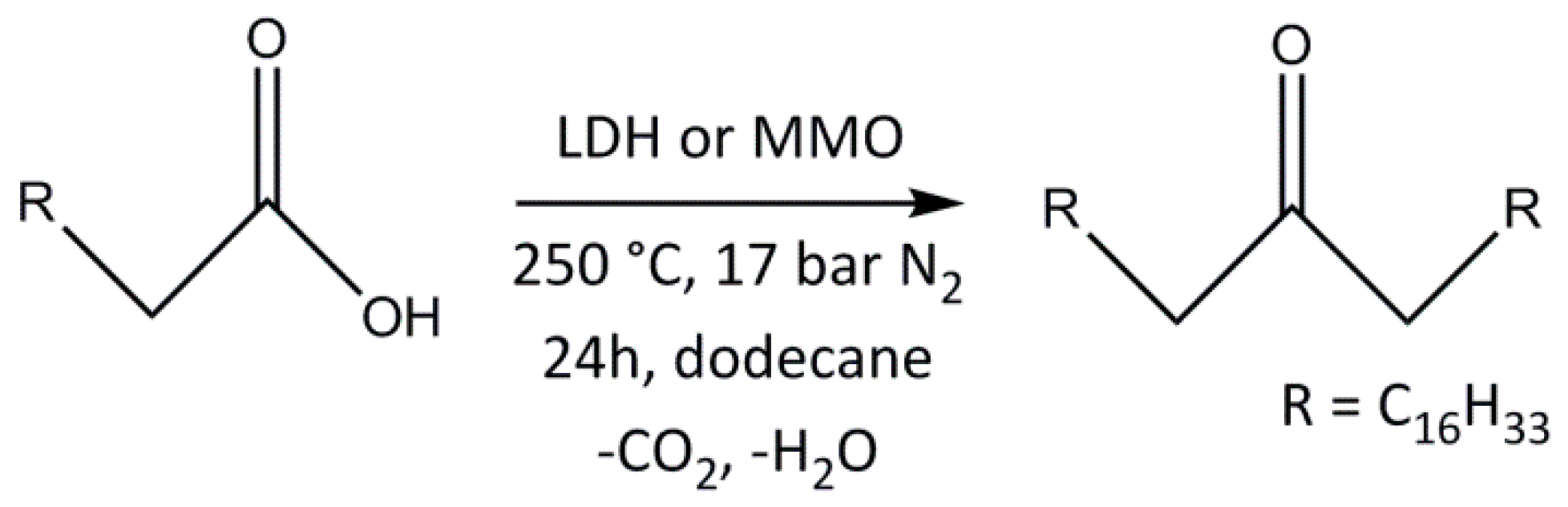
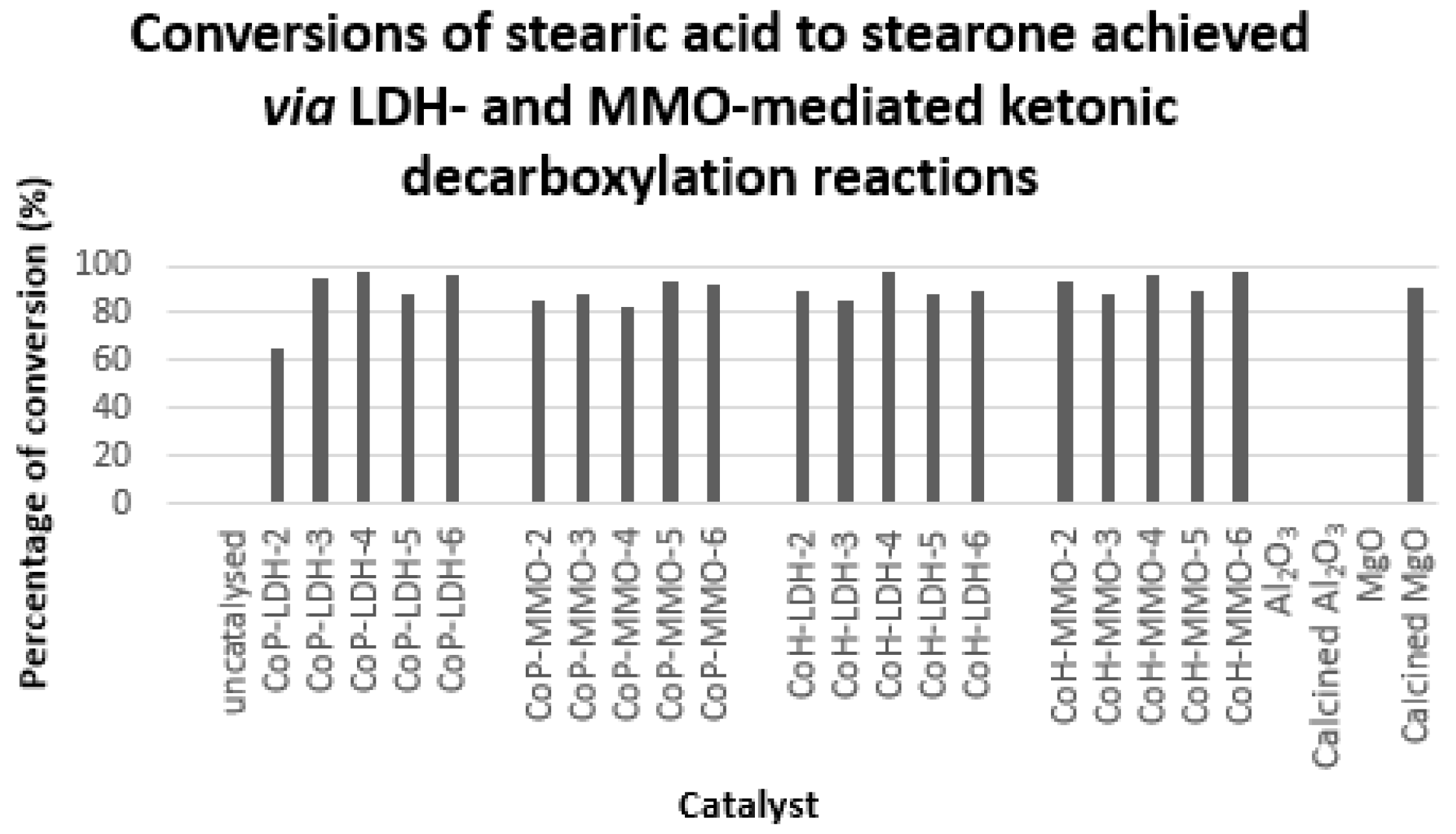
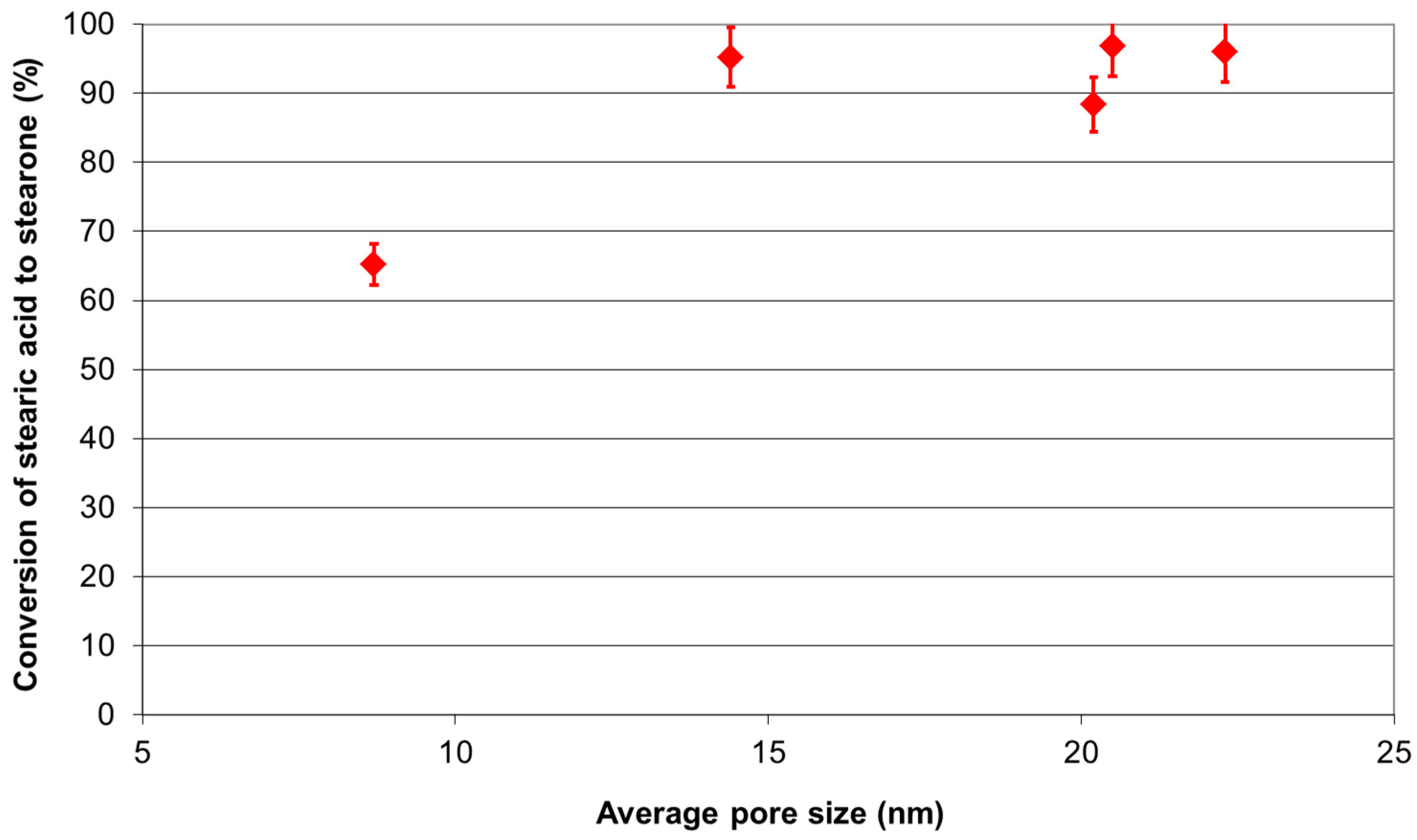
2.2. Analysis of Catalytic Ketonic Decarboxylation Reactions
Ketonic Decarboxylation of Stearic Acid
3. Discussion
3.1. Structure of Mixed Metal Oxide and Layered Double Hydroxide Catalysts
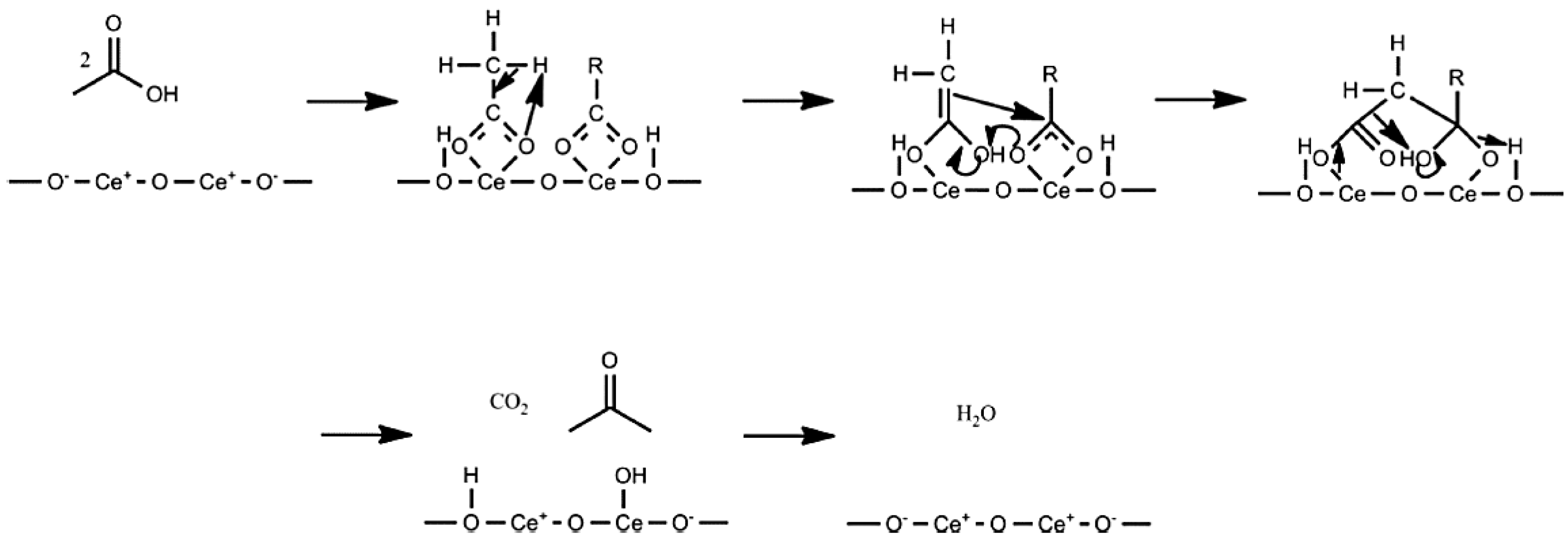
3.2. Ketonic Decarboxylation Reactions
3.3. Comparison of Ketonic Decarboxylation by Mixed Metal Oxides versus Layered Double Hydroxides
3.4. Comparison of Ketonic Decarboxylation by Co-hydrated and Co-precipitated Catalysts as a Function of Mg/Al Ratio
4. Materials and Methods
4.1. Sample Nomenclature
4.2. Catalyst Preparation
4.2.1. Preparation of Layered Double Hydroxides by Co-Precipitation
4.2.2. Preparation of Layered Double Hydroxides via Co-Hydration
4.2.3. Mixed Metal Oxide Preparation
4.3. Material Characterisation
4.3.1. Powder X-ray Diffraction
4.3.2. Thermal Analysis
4.3.3. Scanning Electron Microscopy (SEM)
4.3.4. Inductively Coupled Plasma Optical Emission Spectroscopy
4.3.5. Surface Area Analysis
4.3.6. Estimation of basicity
4.4. Stearic Acid Ketonic Decarboxylation Studies
4.5. Product Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Demirbas, A. Political, Economic and Environmental Impacts of Biofuels: A Review. Appl. Energy 2009, 86, S108–S117. [Google Scholar] [CrossRef]
- Roddy, D.J. Biomass in a Petrochemical World. Interface Focus 2013, 3, 20120038. [Google Scholar] [CrossRef] [PubMed]
- Speight, J. The Biofuels Handbook; RSC Publishing: London, UK, 2011. [Google Scholar]
- McNeil, J.; Day, P.; Sirovski, F. Glycerine from Biodiesel: The Perfect Diesel Fuel. Process Saf. Environ. Prot. 2012, 90, 180–188. [Google Scholar] [CrossRef]
- Carlos Serrano-Ruiz, J.; Pineda, A.; Mariana Balu, A.; Luque, R.; Manuel Campelo, J.; Angel Romero, A.; Manuel Ramos-Fernandez, J. Catalytic transformations of biomass-derived acids into advanced biofuels. Catal. Today 2012, 195, 162–168. [Google Scholar] [CrossRef]
- Shylesh, S.; Gokhale, A.; Ho, C.; Bell, A. Novel Strategies for the Production of Fuels, Lubricants, and Chemicals from Biomass. Acc. Chem. Res. 2017, 50, 2589–2597. [Google Scholar] [CrossRef] [PubMed]
- Rattanasumrit, A.; Ruangpornvisuti, V. Theoretical Study of Conversion Reactions of Ketone to Hydroxyalkylene in Cluster Models of Zeolite H-ZSM-5. J. Mol. Catal. A Chem. 2005, 239, 68–75. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, S.; Zhu, Y.; Li, X.; Luo, Z. Catalytic Cracking of Ketone Components in Biomass Pyrolysis Oil. In Proceedings of the 2010 Asia-Pacific Power and Energy Engineering Conference (APPEEC), Chengdu, China, 28–31 March 2010; pp. 1–3. [Google Scholar]
- Snell, R.W.; Shanks, B.H. Insights into the Ceria-Catalyzed Ketonization Reaction for Biofuels Applications. ACS Catal. 2013, 3, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Siegel, H.; Manfred, E. Ketones. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- Deng, L.; Fu, Y.; Guo, Q.-X. Upgraded Acidic Components of Bio-oil through Catalytic Ketonic Condensation. Energy Fuels 2009, 23, 564–568. [Google Scholar] [CrossRef]
- Nagashima, O.; Sato, S.; Takahashi, R.; Sodesawa, T. Ketonization of carboxylic acids over CeO2-based composite oxides. J. Mol. Catal. A Chem. 2005, 227, 231–239. [Google Scholar] [CrossRef]
- Shutilov, A.A.; Simonov, M.N.; Zaytseva, Y.A. Phase composition and catalytic properties of ZrO2 and CeO2-ZrO2 in the ketonization of pentanoic acid to 5-nonanone. Kinetics Catal. 2013, 54, 184–192. [Google Scholar] [CrossRef]
- Gaertner, C.A.; Serrano-Ruiz, J.C.; Braden, D.J.; Dumesic, J.A. Catalytic Coupling of Carboxylic Acids by Ketonization as a Processing Step in Biomass Conversion. J. Catal. 2009, 266, 71–78. [Google Scholar] [CrossRef]
- Yamada, Y.; Segawa, M.; Sato, F.; Kojima, T.; Sato, S. Catalytic Performance of Rare Earth Oxides in Ketonization of Acetic Acid. J. Mol. Catal. A Chem. 2011, 346, 79–86. [Google Scholar] [CrossRef]
- Jackson, M.A.; Cermak, S.C. Cross Ketonization of Cuphea sp Oil with Acetic Acid over a Composite Oxide of Fe, Ce, and Al. Appl. Catal. A Gen. 2012, 431, 157–163. [Google Scholar] [CrossRef]
- Pestman, R.; Koster, R.M.; van Duijne, A.; Pieterse, J.A.Z.; Ponec, V. Reactions of Carboxylic Acids on Oxides: 2. Bimolecular Reaction of Aliphatic Acids to Ketones. J. Catal. 1997, 168, 265–272. [Google Scholar] [CrossRef]
- Corma, A.; Borja, O.T.; Renz, M.; Simakova, I.L. Conversion of levulinic acid derived valeric acid into a liquid transportation fuel of the kerosene type. J. Mol. Catal. A Chem. 2014, 388–389 (Suppl. C), 116–122. [Google Scholar] [CrossRef]
- Pham, T.N.; Sooknoi, T.; Crossley, S.P.; Resasco, D.E. Ketonization of Carboxylic Acids: Mechanisms, Catalysts, and Implications for Biomass Conversion. ACS Catal. 2013, 3, 2456–2473. [Google Scholar] [CrossRef]
- Pacchioni, G. Ketonization of Carboxylic Acids in Biomass Conversion over TiO2 and ZrO2 Surfaces: A DFT Perspective. ACS Catal. 2014, 4, 2874–2888. [Google Scholar] [CrossRef]
- Neunhoeffer, O.; Paschke, P. Über den Mechanismus der Ketonbildung aus Carbonsäuren. Berichte der deutschen chemischen Gesellschaft 1939, 72, 919–929. [Google Scholar] [CrossRef]
- Ignatchenko, A.V. Density Functional Theory Study of Carboxylic Acids Adsorption and Enolization on Monoclinic Zirconia Surfaces. J. Phys. Chem. C 2011, 115, 16012–16018. [Google Scholar] [CrossRef]
- Ignatchenko, A.V.; Kozliak, E.I. Distinguishing Enolic and Carbonyl Components in the Mechanism of Carboxylic Acid Ketonization on Monoclinic Zirconia. ACS Catal. 2012, 2, 1555–1562. [Google Scholar] [CrossRef]
- Randery, S.D.; Warren, J.S.; Dooley, K.M. Cerium Oxide-based Catalysts for Production of Ketones by Acid Condensation. Appl. Catal. A Gen. 2002, 226, 265–280. [Google Scholar] [CrossRef]
- Pham, T.N.; Shi, D.; Resasco, D.E. Kinetics and Mechanism of Ketonization of Acetic Acid on Ru/TiO2 Catalyst. Top. Catal. 2014, 57, 706–714. [Google Scholar] [CrossRef]
- Pulido, A.; Oliver-Tomas, B.; Renz, M.; Boronat, M.; Corma, A. Ketonic Decarboxylation Reaction Mechanism: A Combined Experimental and DFT Study. ChemSusChem 2013, 6, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Enjamuri, N.; Shah, S.; Sadeq Al-Fatesh, A.; Bravo-Suárez, J.J.; Chowdhury, B. Ketonization of Oxygenated Hydrocarbons on Metal Cxide based Catalysts. Catal. Today 2018, 302, 16–49. [Google Scholar] [CrossRef]
- Evans, D.G.; Slade, R.C.T. Structural Aspects of Layered Double Hydroxides. Struct. Bond. 2006, 119, 1–87. [Google Scholar]
- Wang, Q.; O’Hare, D. Recent Advances in the Synthesis and Application of Layered Double Hydroxide (LDH) Nanosheets. Chem. Rev. 2012, 112, 4124–4155. [Google Scholar] [CrossRef] [PubMed]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type Anionic Clays: Preparation, Properties and Applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Debecker, D.P.; Gaigneaux, E.M.; Busca, G. Exploring, Tuning, and Exploiting the Basicity of Hydrotalcites for Applications in Heterogeneous Catalysis. Chem. A Eur. J. 2009, 15, 3920–3935. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Kumura, T. Synthesis of New Hydrotalcite-like Compounds and Their Physicochemical Properties. Chem. Lett. 1973, 2, 843–848. [Google Scholar] [CrossRef]
- Costantino, U.; Marmottini, F.; Nocchetti, M.; Vivani, R. New Synthetic Routes to Hydrotalcite-like Compounds—Characterisation and Properties of the Obtained Materials. Eur. J. Inorg. Chem. 1998, 10, 1439–1446. [Google Scholar] [CrossRef]
- Greenwell, H.C.; Jones, W.; Stamires, D.N.; O’Connor, P.; Brady, M.F. A One-pot Synthesis of Hybrid Organo-layered Double Hydroxide Catalyst Precursors. Green Chem. 2006, 8, 1067–1072. [Google Scholar] [CrossRef]
- Greenwell, H.C.; Jones, W.; Rugen-Hankey, S.L.; Holliman, P.J.; Thompson, R.L. Efficient Synthesis of Ordered Organo-layered Double Hydroxides. Green Chem. 2010, 12, 688–695. [Google Scholar] [CrossRef]
- Greenwell, H.C.; Marsden, C.C.; Jones, W. Synthesis of Organo-layered Double Hydroxides by an Environmentally Friendly Co-Hydration Route. Green Chem. 2007, 9, 1299–1307. [Google Scholar] [CrossRef]
- Kagunya, W.; Hassan, Z.; Jones, W. Catalytic Properties of Layered Double Hydroxides and Their Calcined Derivatives. Inorg. Chem. 1996, 35, 5970–5974. [Google Scholar] [CrossRef]
- Hernandez, M.; Lauwaert, J.; Van Der Voort, P. Recent Advances on the Utilization of Layered Double Hydroxides (LDHs) and Related Heterogeneous Catalysts in a Lignocellulosic-Feedstock Biorefinery Scheme. Green Chem. 2017, 19, 5259–5516. [Google Scholar] [CrossRef]
- Yan, K.; Liu, Y.; Lu, Y.; Chai, J.; Sun, L. Catalytic application of layered double hydroxide-derived catalyst for the conversion of biomass-derived molecules. Catal. Sci. Technol. 2017, 7, 1622–1645. [Google Scholar] [CrossRef]
- Roeffaers, M.B.J.; Sels, B.F.; Uji-i, H.; De Schryver, F.C.; Jacobs, P.A.; De Vos, D.E.; Hofkens, J. Spatially Resolved Observation of Crystal-Face-Dependent Catalysis by Single Turnover Counting. Nature 2006, 439, 572–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueras, F.O. Base Catalysis in the Synthesis of Fine Chemicals. Top. Catal. 2004, 29, 189–196. [Google Scholar] [CrossRef]
- Lee, D.; Park, Y.; Lee, K. Heterogeneous Base Catalysts for Transesterification in Biodiesel Synthesis. Catal. Surv. Asia 2009, 13, 63–77. [Google Scholar] [CrossRef]
- Goswamee, R.L.; Saikia, J.; Allou, N.B. Use of Calcined Mg–Al Layered Double Hydroxides to Regulate Endocrine Disruptor Methylparaben in Excess as Adsorbent and as Control Releasing Agent in Normal Situations. J. Environ. Chem. Eng. 2017, 6, 1189–1200. [Google Scholar]
- Guida, A.; Lhouty, M.H.; Tichit, D.; Figueras, F.; Geneste, P. Hydrotalcites as Base Catalysts. Kinetics of Claisen-Schmidt Condensation, Intramolecular Condensation of Acetonylacetone and Synthesis of Chalcone. Appl. Catal. A Gen. 1997, 164, 251–264. [Google Scholar] [CrossRef]
- Brito, A.; Borges, M.E.; Garin, M.; Hernandez, A. Biodiesel Production from Waste Oil Using Mg–Al Layered Double Hydroxide Catalysts. Energy Fuels 2009, 23, 2952–2958. [Google Scholar] [CrossRef]
- Choudary, B.M.; Kantam, M.L.; Reddy, C.R.V.; Rao, K.K.; Figueras, F. The first example of Michael addition catalysed by modified Mg–Al hydrotalcite. J. Mol. Catal. A Chem. 1999, 146, 279–284. [Google Scholar] [CrossRef]
- Parida, K.; Das, J. Mg/Al Hydrotalcites: Preparation, Characterisation and Ketonisation of Acetic Acid. J. Mol. Catal. A Chem. 2000, 151, 185–192. [Google Scholar] [CrossRef]
- Constantino, V.R.L.; Pinnavaia, T.J. Basic Properties of Mg1−X2+AlX3+ Layered Double Hydroxides Intercalated by Carbonate, Hydroxide, Chloride and Sulfate Aanions. Inorg. Chem. 1995, 34, 883–892. [Google Scholar] [CrossRef]
- Perez-Ramirez, J.; Abello, S.; van der Pers, N.M. Influence of the divalent cation on the thermal activation and reconstruction of hydrotalcite-like compounds. J. Phys. Chem. C 2007, 111, 3642–3650. [Google Scholar] [CrossRef]
- Tichit, D.; Gerardin, C.; Durand, R.; Coq, B. Layered Double Hydroxides: Precursors for Multifunctional Catalysts. Top. Catal. 2006, 39, 89–96. [Google Scholar] [CrossRef]
- Constantino, V.R.L.; Pinnavaia, T.J. Structure-Reactivity Relationships for Basic Catalysts Derived from a Mg2+Al3+(CO32−) Layered Double Hydroxide. Catal. Lett. 1994, 23, 361–367. [Google Scholar] [CrossRef]
- Cantrell, D.G.; Gillie, L.J.; Lee, A.F.; Wilson, K. Structure-reactivity Correlations in MgAl Hydrotalcite Catalysts for Biodiesel Synthesis. Appl. Catal. A Gen. 2005, 287, 183–190. [Google Scholar] [CrossRef]
- Cross, H.E.; Brown, D.R. Entrained Sodium in Mixed Metal Oxide Catalysts Derived from Layered Double Hydroxides. Catal. Commun. 2010, 12, 243–245. [Google Scholar] [CrossRef]
- He, J.; Wei, M.; Li, B.; Kang, Y.; Evans, D.G.; Duan, X. Preparation of Layered Double Hydroxides. In Layered Double Hydroxides; Duan, X., Evans, D.G., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 89–119. [Google Scholar]
- Tichit, D.; Coq, B. Catalysis by Hydrotalcites and Related Materials. Cattech 2003, 7, 206–217. [Google Scholar] [CrossRef]
- Hammett, L.P.; Deyrup, A.J. A series of Simple Basic Indicators. I. The Acidity Functions of Mixtures of Sulfuric and Perchloric Acids with Water. J. Am. Chem. Soc. 1932, 54, 2721–2739. [Google Scholar] [CrossRef]
- Xie, W.L.; Peng, H.; Chen, L.G. Calcined Mg–Al Hydrotalcites as Solid Base Catalysts for Methanolysis of Soybean Oil. J. Mol. Catal. A Chem. 2006, 246, 24–32. [Google Scholar] [CrossRef]
- Prinetto, F.; Ghiotti, G.; Durand, R.; Tichit, D. Investigation of Acid-Base Properties of Catalysts Obtained from Layered Double Hydroxides. J. Phys. Chem. B 2000, 104, 11117–11126. [Google Scholar] [CrossRef]
- Shen, J.Y.; Kobe, J.M.; Chen, Y.; Dumesic, J.A. Synthesis and Surface Acid/Base Properties of Magnesium–Aluminum Mixed Oxides Obtained from Hydrotalcites. Langmuir 1994, 10, 3902–3908. [Google Scholar] [CrossRef]
- Hobbs, C.; Jaskaniec, S.; McCarthy, E.K.; Downing, C.; Opelt, K.; Güth, K.; Shmeliov, A.; Mourad, M.C.D.; Mandel, K.; Nicolosi, V. Structural Transformation of Layered Double Hydroxides: An in situ TEM Analysis. 2D Mater. Appl. 2018, 2, 4. [Google Scholar] [CrossRef]
- Mahjoubi, F.Z.; Khalidi, A.; Abdennouri, M.; Barka, N. Zn–Al Layered Double Hydroxides Intercalated with Carbonate, Nitrate, Chloride and Sulphate Ions: Synthesis, Characterisation and Dye Removal Properties. J. Taibah Univ. Sci. 2017, 11, 90–100. [Google Scholar] [CrossRef]
- Kaneyoshi, M.; Jones, W. Formation of Mg–Al Layered Double Hydroxides Intercalated with Nitrilotriacetate Anions. J. Mater. Chem. 1999, 9, 805–811. [Google Scholar] [CrossRef]
- Hudson, M.J.; Carlino, S.; Apperley, D.C. Thermal Conversion of a Layered (Mg/Al) Double Hydroxide to the Oxide. J. Mater. Chem. 1995, 5, 323–329. [Google Scholar] [CrossRef]
- Li, S.-S.; Jiang, M.; Jiang, T.-J.; Liu, J.-H.; Guo, Z.; Huang, X.-J. Competitive Adsorption Behavior Toward Metal Ions On Nano-Fe/Mg/Ni Ternary Layered Double Hydroxide Proved by XPS: Evidence of Selective and Sensitive Detection of Pb(II). J. Hazardous Mater. 2017, 338, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Valente, J.S.; Sanchez-Cantu, M.; Lima, E.; Figueras, F. Method for Large-Scale Production of Multimetallic Layered Double Hydroxides: Formation Mechanism Discernment. Chem. Mater. 2009, 21, 5809–5818. [Google Scholar] [CrossRef]
- Rives, V. Layered Double Hydroxides: Present and Future; Nova Science Publishers: Hauppauge, NY, USA, 2001. [Google Scholar]
- Reichle, W.T.; Kang, S.Y.; Everhardt, D.S. The Nature of the Thermal-Decomposition of a Catalytically Active Anionic Clay Mineral. J. Catal. 1986, 101, 352–359. [Google Scholar] [CrossRef]
- Velu, S.; Suzuki, K.; Okazaki, M.; Osaki, T.; Tomura, S.; Ohashi, F. Synthesis of New Sn-incorporated Layered Double Hydroxides and their Thermal Evolution to Mixed Oxides. Chem. Mater. 1999, 11, 2163–2172. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, F.; Zhang, R.; Evans, D.G.; Duan, X. Preparation of Layered Double-Hydroxide Nanomaterials with a Uniform Crystallite Size Using a New Method Involving Separate Nucleation and Aging Steps. Chem. Mater. 2002, 14, 4286–4291. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Reporting Physisorption Data for Gas Solid Systems with Special Reference to the Determination of Surface-Area and Porosity. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Kang, D.; Yu, X.; Tong, S.; Ge, M.; Zuo, J.; Cao, C.; Song, W. Performance and Mechanism of Mg/Fe Layered Double Hydroxides for Fluoride and Arsenate Removal from Aqueous Solution. Chem. Eng. J. 2013, 228, 731–740. [Google Scholar] [CrossRef]
- Greenwell, H.C.; Stackhouse, S.; Coveney, P.V.; Jones, W. A Density Functional Theory Study of Catalytic Trans-Esterification by Tert-Butoxide Mgal Anionic Clays. J. Phys. Chem. B 2003, 107, 3476–3485. [Google Scholar] [CrossRef]
- Shichi, T.; Takagi, K.; Sawaki, Y. Stereoselectivity Control of 2 + 2 Photocycloaddition by Changing Site Distances of Hydrotalcite Interlayers. Chem. Commun. 1996, 17, 2027–2028. [Google Scholar] [CrossRef]
- Takagi, K.; Shichi, T.; Usami, H.; Sawaki, Y. Controlled Photocycloaddition of Unsaturated Carboxylates Intercalated in Hydrotalcite Clay Interlayers. J. Am. Chem. Soc. 1993, 115, 4339–4344. [Google Scholar] [CrossRef]
- Zhang, A.H.; Ma, Q.S.; Wang, K.S.; Liu, X.C.; Shuler, P.; Tang, Y.C. Naphthenic Acid Removal from Crude Oil Through Catalytic Decarboxylation On Magnesium Oxide. Appl. Catal. A Gen. 2006, 303, 103–109. [Google Scholar] [CrossRef]
- Corma, A.; Renz, M.; Schaverien, C. Coupling Fatty Acids by Ketonic Decarboxylation Using Solid Catalysts for the Direct Production of Diesel, Lubricants, and Chemicals. ChemSusChem 2008, 1, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Verziu, M.; Cojocaru, B.; Hu, J.; Richards, R.; Ciuculescu, C.; Filip, P.; Parvulescu, V.I. Sunflower and Rapeseed Oil Transesterification to Biodiesel Over Different Nanocrystalline MgO Catalysts. Green Chem. 2008, 10, 373–381. [Google Scholar] [CrossRef]
- Na, J.G.; Yi, B.E.; Kim, J.N.; Yi, K.B.; Park, S.Y.; Park, J.H.; Ko, C.H. Hydrocarbon Production from Decarboxylation of Fatty Acid Without Hydrogen. Catal. Today 2010, 156, 44–48. [Google Scholar] [CrossRef]
- Gaertner, C.A.; Serrano-Ruiz, J.C.; Braden, D.J.; Dumesic, J.A. Ketonization Reactions of Carboxylic Acids and Esters over Ceria–Zirconia as Biomass-Upgrading Processes. Ind. Eng. Chem. Res. 2010, 49, 6027–6033. [Google Scholar] [CrossRef]
- Kubičková, I.; Snåre, M.; Eränen, K.; Mäki-Arvela, P.; Murzin, D. Hydrocarbons for diesel fuel via decarboxylation of vegetable oils. Catal. Today 2005, 106, 197–200. [Google Scholar] [CrossRef]
- Anastas, P.; Warner, J. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Clarity Chromatography Station, Clarity Chromatography Station version 2.4.1.57; DataApex: Prague, Czech Republic, 2003.
- ASTM D6584-10ae1, Standard Test Method for Determination of Total Monoglyceride, Total Diglyceride, Total Triglyceride, and Free and Total Glycerin in B-100 Biodiesel Methyl Esters by Gas Chromatography; ASTM International: West Conshohocken, PA, USA, 2010.
- Barthomeuf, D. Conjugate Acid-base Pairs in Zeolites. J. Phys. Chem. 1984, 88, 42–45. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, M.; Blasco, T. Characterization of Zeolite Basicity Using Probe Molecules by Means of Infrared and Solid State NMR Spectroscopies. Catal. Today 2009, 143, 293–301. [Google Scholar] [CrossRef]
- Griinert, W.; Muhler, M.; Schroder, K.; Sauer, J.; Schloegl, R. Investigations of Zeolites by Photoelectron and Ion Scattering Spectroscopy. 2. A New Interpretation of XPS Binding Energy Shifts in Zeolites. J. Phys. Chem. 1994, 98, 10920–10929. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a/Å | c/Å | Average Crystal Size a/nm | Average Crystal Size c/nm | PercentageAl % | Expected Ratio of Mg:Al | ICP Ratio of Mg:Al | Surface Area/m2·g−1 | Pore Volume/cm3·g−1 | Average Pore Size/nm | Surface Basicity/pH |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CoP-LDH-2 | 3.05 (0.15) | 22.94 (1.14) | 255 (13) | 221 (11) | 31.9 (1.6) | 2.0 | 1.7 (0.03) | 91 | 0.32 | 9 | 9.0–10.0 |
| CoP-LDH-3 | 3.06 (0.15) | 23.42 (1.18) | 198 (10) | 129 (6) | 25.1 (1.3) | 3.0 | 2.7 (0.03) | 82 | 0.42 | 14 | 7.6–9.0 |
| CoP-LDH-4 | 3.07 (0.15) | 23.65 (1.18) | 254 (13) | 133 (7) | 20.2 (1.0) | 4.0 | 3.3 (0.03) | 17 | 0.12 | 21 | 9.0–10.0 |
| CoP-LDH-5 | 3.08 (0.15) | 23.83 (1.19) | 168 (8) | 114 (6) | 18.3 (0.9) | 5.0 | 4.0 (0.03) | 24 | 0.17 | 20 | 9.0–10.0 |
| CoP-LDH-6 | 3.08 (0.15) | 23.82 (1.19) | 161 (8) | 91 (5) | 18.0 (0.9) | 6.0 | 5.2 (0.03) | 39 | 0.25 | 22 | 9.0–10.0 |
| CoH-LDH-2 | 3.14 (0.16) | 23.50 (1.18) | 312 (16) | 140 (7) | - | 2.0 | 1.3 (0.03) | 33 | 0.05 | 10 | 6.0–7.6 |
| CoH-LDH-3 | 3.14 (0.16) | 23.93 (1.20) | 301 (15) | 102 (5) | - | 3.0 | 2.3 (0.03) | 42 | 0.08 | 10 | 6.0–7.6 |
| CoH-LDH-4 | 3.14 (0.16) | 24.26 (1.21) | 289 (14) | 138 (7) | - | 4.0 | 3.7 (0.03) | 46 | 0.10 | 11 | 6.0–7.6 |
| CoH-LDH-5 | 3.14 (0.16) | 23.98 (1.20) | 387 (19) | 116 (6) | - | 5.0 | 3.2 (0.03) | 42 | 0.09 | 13 | 7.6–9.0 |
| CoH-LDH-6 | 3.14 (0.16) | 23.86 (1.19) | 337 (17) | 189 (9) | - | 6.0 | 5.0 (0.03) | 43 | 0.10 | 11 | 7.6–9.0 |
| Sample | Surface Area/m2·g−1 | Pore Volume/cm3·g−1 | Average Pore Size/nm | Surface Basicity (pH) |
|---|---|---|---|---|
| CoP-MMO-2 | 163 | 0.53 | 10 | 7.6–9.0 |
| CoP-MMO-3 | 155 | 0.61 | 15 | 7.6–9.0 |
| CoP-MMO-4 | 190 | 0.40 | 6 | 9.0–10.0 |
| CoP-MMO-5 | 199 | 0.49 | 7 | 9.0–10.0 |
| CoP-MMO-6 | 160 | 0.49 | 9 | 7.6–9.0 |
| CoH-MMO-2 | 231 | 0.32 | 5 | 7.6–9.0 |
| CoH-MMO-3 | 213 | 0.39 | 6 | 7.6–9.0 |
| CoH-MMO-4 | 184 | 0.37 | 6 | 7.6–9.0 |
| CoH-MMO-5 | 156 | 0.37 | 8 | 7.6–9.0 |
| CoH-MMO-6 | 198 | 0.39 | 6 | 7.6–9.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, B.; Li, L.; Perera-Solis, D.D.; Gildea, L.F.; Zholobenko, V.L.; Dyer, P.W.; Greenwell, H.C. Ketone Formation via Decarboxylation Reactions of Fatty Acids Using Solid Hydroxide/Oxide Catalysts. Inorganics 2018, 6, 121. https://doi.org/10.3390/inorganics6040121
Smith B, Li L, Perera-Solis DD, Gildea LF, Zholobenko VL, Dyer PW, Greenwell HC. Ketone Formation via Decarboxylation Reactions of Fatty Acids Using Solid Hydroxide/Oxide Catalysts. Inorganics. 2018; 6(4):121. https://doi.org/10.3390/inorganics6040121
Chicago/Turabian StyleSmith, Benjamin, Li Li, Diego D. Perera-Solis, Louise F. Gildea, Vladimir L. Zholobenko, Philip W. Dyer, and H. Christopher Greenwell. 2018. "Ketone Formation via Decarboxylation Reactions of Fatty Acids Using Solid Hydroxide/Oxide Catalysts" Inorganics 6, no. 4: 121. https://doi.org/10.3390/inorganics6040121
APA StyleSmith, B., Li, L., Perera-Solis, D. D., Gildea, L. F., Zholobenko, V. L., Dyer, P. W., & Greenwell, H. C. (2018). Ketone Formation via Decarboxylation Reactions of Fatty Acids Using Solid Hydroxide/Oxide Catalysts. Inorganics, 6(4), 121. https://doi.org/10.3390/inorganics6040121