Gold(III) Pyridine-Benzimidazole Complexes as Aquaglyceroporin Inhibitors and Antiproliferative Agents

 , , , ,
, , , ,  and
and
Abstract
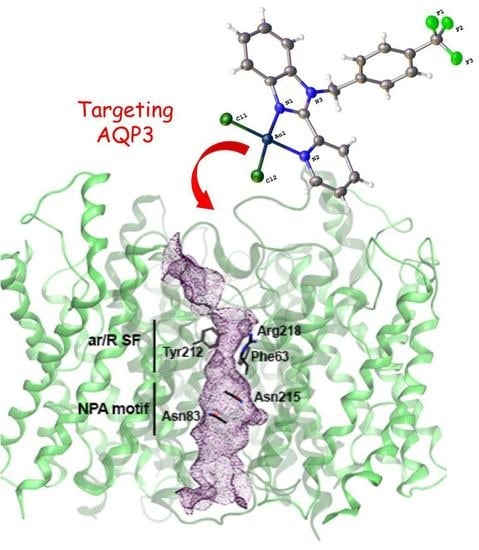
:
1. Introduction
2. Results
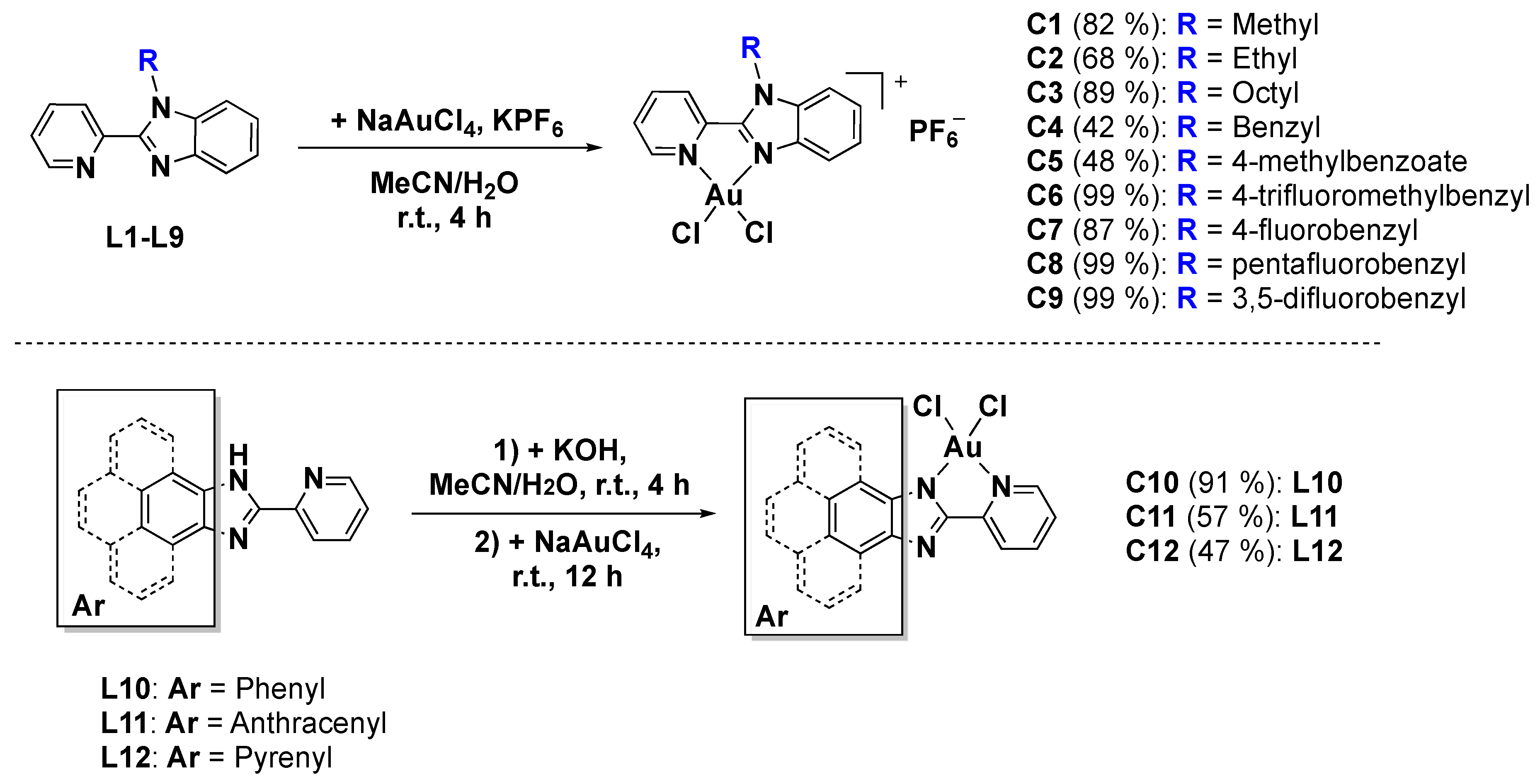
2.1. Synthesis and Characterization of Au(III) Complexes
2.2. UV–Visible Stability Studies
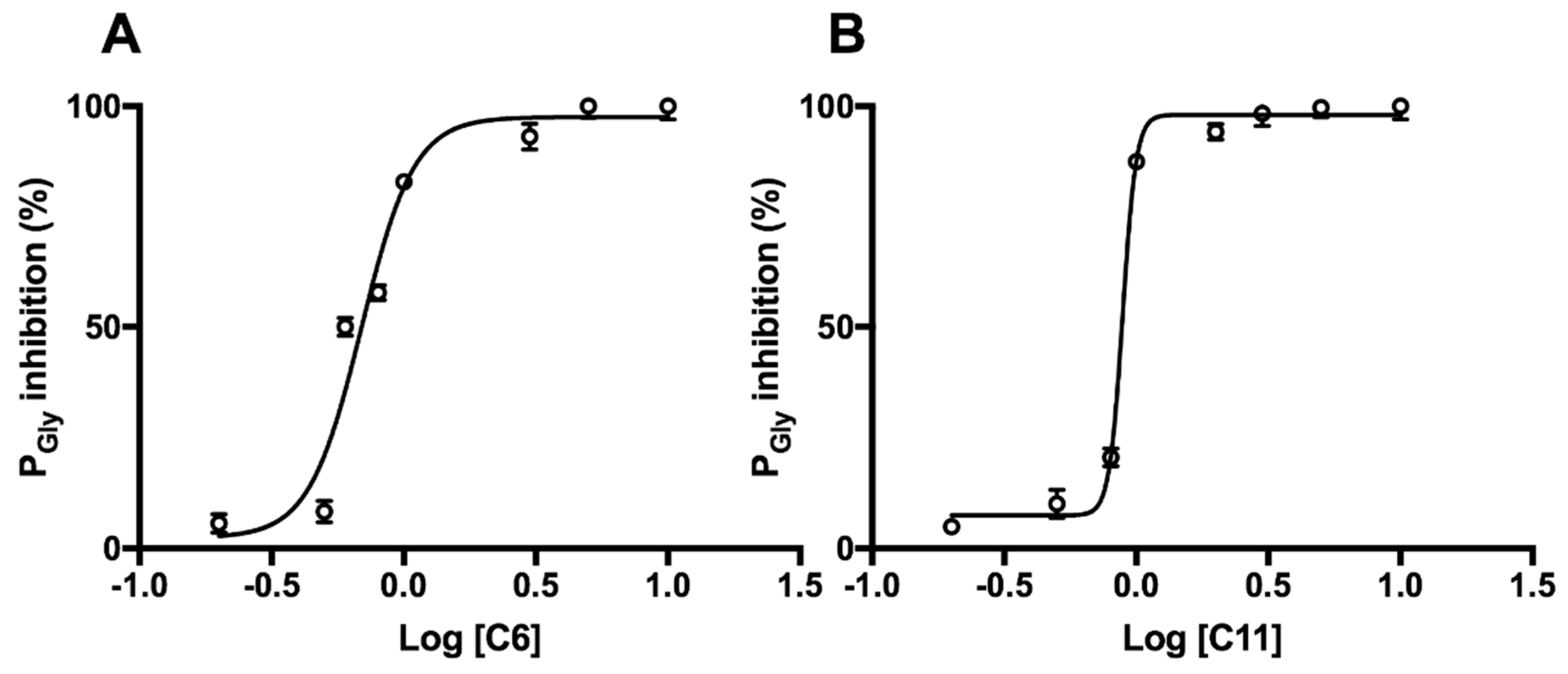
2.3. Inhibition of Aquaporins
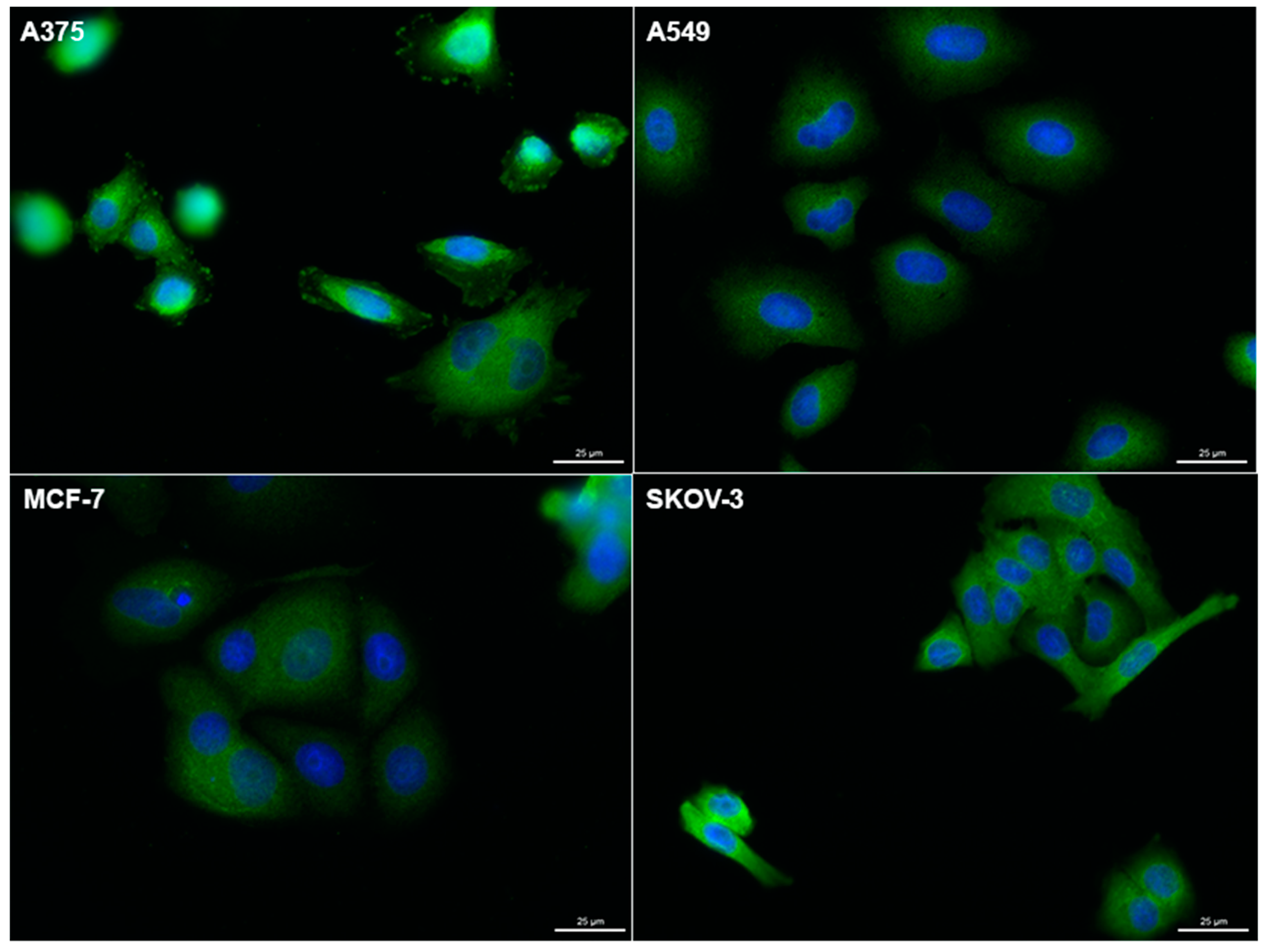
2.4. Expression of AQP3 in Cancer Cells and Antiproliferative Activities
3. Materials and Methods
3.1. General Information
3.2. Compounds Synthesis
3.2.1. General Procedure for the Synthesis of Ligands L1–L9
3.2.2. General Procedure for the Synthesis of the Cationic Complexes C1–C9
3.2.3. General Procedure for the Synthesis of the Neutral Complexes C10–C12
3.3. UV–Visible and Fluorescence Spectroscopy
3.4. X-ray Diffraction Analysis
3.5. UV–Visible Absorption Spectroscopy
3.6. Aquaporins Inhibition
3.7. Cell Lines and Culture Conditions
3.8. Immunocytofluorescence
3.9. Flow Cytometry Analysis
3.10. Antiproliferative Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Casini, A.; Sun, R.W.-Y.; Ott, I. Medicinal Chemistry of Gold Anticancer Metallodrugs. In Metallo-Drugs: Development and Action of Anticancer Agents; Sigel, A., Sigel, H., Freisinger, E., Sigel, R.K.O., Eds.; De Gruyter: Berlin, Germany; Boston, MA, USA, 2018; pp. 199–217. [Google Scholar]
- Shaw, C.F. Gold-based therapeutic agents. Chem. Rev. 1999, 99, 2589–2600. [Google Scholar] [CrossRef]
- De Almeida, A.; Oliveira, B.L.; Correia, J.D.G.; Soveral, G.; Casini, A. Emerging protein targets for metal-based pharmaceutical agents: An update. Coord. Chem. Rev. 2013, 257, 2689–2704. [Google Scholar] [CrossRef]
- de Almeida, A.; Soveral, G.; Casini, A. Gold compounds as aquaporin inhibitors: New opportunities for therapy and imaging. Med. Chem. Commun. 2014, 5, 1444–1453. [Google Scholar] [CrossRef]
- Agre, P. Aquaporin Water Channels (Nobel Lecture). Angew. Chem. Int. Ed. 2004, 43, 4278–4290. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci. 2005, 118, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Soveral, G.; Nielsen, S.; Casini, A. Aquaporins in Health and Disease: New Molecular Targets for Drug Discovery, 1st ed.; Soveral, G., Nielsen, S., Casini, A., Eds.; CRC Press: Boca Raton, FL, USA, 2016; ISBN 978-971-4987-0783-1. [Google Scholar]
- Madeira, A.; Fernández-Veledo, S.; Camps, M.; Zorzano, A.; Moura, T.F.; Ceperuelo-Mallafré, V.; Vendrell, J.; Soveral, G. Human Aquaporin-11 is a water and glycerol channel and localizes in the vicinity of lipid droplets in human adipocytes. Obesity 2014, 22, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Tanaka, Y.; Morishita, Y. The role of mammalian superaquaporins inside the cell. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Aikman, B.; De Almeida, A.; Meier-Menches, S.M.; Casini, A. Aquaporins in cancer development: Opportunities for bioinorganic chemistry to contribute novel chemical probes and therapeutic agents. Metallomics 2018, 10, 696–712. [Google Scholar] [CrossRef] [PubMed]
- Soveral, G.; Casini, A. Aquaporin modulators: A patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.P.; Marrone, A.; Ciancetta, A.; Cobo, A.G.; Echevarría, M.; Moura, T.F.; Re, N.; Casini, A.; Soveral, G. Targeting aquaporin function: Potent inhibition of aquaglyceroporin-3 by a gold-based compound. PLoS ONE 2012, 7, e37435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.P.; Ciancetta, A.; deAlmeida, A.; Marrone, A.; Re, N.; Soveral, G.; Casini, A. Aquaporin inhibition by gold(III) compounds: New insights. ChemMedChem 2013, 8, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Serna, A.; Galán-Cobo, A.; Rodrigues, C.; Sánchez-Gomar, I.; Toledo-Aral, J.J.; Moura, T.F.; Casini, A.; Soveral, G.; Echevarría, M. Functional Inhibition of Aquaporin-3 With a Gold-Based Compound Induces Blockage of Cell Proliferation. J. Cell. Physiol. 2014, 229, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.; Mósca, A.F.; Wragg, D.; Wenzel, M.; Kavanagh, P.; Barone, G.; Leoni, S.; Soveral, G.; Casini, A. The mechanism of aquaporin inhibition by gold compounds elucidated by biophysical and computational methods. Chem. Commun. 2017, 53, 3830–3833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, M.; Ryan, A.J.; Little, P.J. Inhibition of rat hepatic microsomal aminopyrine N-demethylase activity by benzimidazole derivatives. Quantitative structure-activity relationships. J. Med. Chem. 1982, 25, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Sontakke, V.A.; Ghosh, S.; Lawande, P.P.; Chopade, B.A.; Shinde, V.S. A simple, efficient synthesis of 2-aryl benzimidazoles using silica supported periodic acid catalyst and evaluation of anticancer activity. ISRN Org. Chem. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Prosser, K.E.; Chang, S.W.; Saraci, F.; Le, P.H.; Walsby, C.J. Anticancer copper pyridine benzimidazole complexes: ROS generation, biomolecule interactions, and cytotoxicity. J. Inorg. Biochem. 2017, 167, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Serratrice, M.; Cinellu, M.A.; Maiore, L.; Pilo, M.; Zucca, A.; Gabbiani, C.; Guerri, A.; Landini, I.; Nobili, S.; Mini, E.; et al. Synthesis, structural characterization, solution behavior, and in vitro antiproliferative properties of a series of gold complexes with 2-(2′-pyridyl)benzimidazole as ligand: Comparisons of gold(III) versus gold(I) and mononuclear versus binuclear derivat. Inorg. Chem. 2012, 51, 3161–3171. [Google Scholar] [CrossRef] [PubMed]
- Gümüş, F.; Pamuk, İ.; Özden, T.; Yıldız, S.; Diril, N.; Öksüzoǧlu, E.; Gür, S.; Özkul, A. Synthesis, characterization and in vitro cytotoxic, mutagenic and antimicrobial activity of platinum(II) complexes with substituted benzimidazole ligands. J. Inorg. Biochem. 2003, 94, 255–262. [Google Scholar] [CrossRef]
- Huang, W.-K.; Cheng, C.-W.; Chang, S.-M.; Lee, Y.-P.; Diau, E.W.-G. Synthesis and electron-transfer properties of benzimidazole-functionalized ruthenium complexes for highly efficient dye-sensitized solar cells. Chem. Commun. 2010, 46, 8992–8994. [Google Scholar] [CrossRef] [PubMed]
- Mardanya, S.; Karmakar, S.; Das, S.; Baitalik, S. Anion and cation triggered modulation of optical properties of a pyridyl-imidazole receptor rigidly linked to pyrene and construction of INHIBIT, OR and XOR molecular logic gates: A combined experimental and DFT/TD-DFT investigation. Sens. Actuators B Chem. 2015, 206, 701–713. [Google Scholar] [CrossRef]
- Petryszak, R.; Keays, M.; Tang, Y.A.; Fonseca, N.A.; Barrera, E.; Burdett, T.; Füllgrabe, A.; Fuentes, A.M.-P.; Jupp, S.; Koskinen, S.; et al. Expression Atlas update—An integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016, 44, D746–D752. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Cao, Q.; Bailie, D.S.; Fu, R.; Muldoon, M.J. Cationic palladium(II) complexes as catalysts for the oxidation of terminal olefins to methyl ketones using hydrogen peroxide. Green Chem. 2015, 17, 2750–2757. [Google Scholar] [CrossRef] [Green Version]
- Sunesh, C.D.; Mathai, G.; Choe, Y. Constructive Effects of Long Alkyl Chains on the Electroluminescent Properties of Cationic Iridium Complex-Based Light-Emitting Electrochemical Cells. ACS Appl. Mater. Interfaces 2014, 6, 17416–17425. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, A.M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef] [Green Version]
- Würth, C.; Grabolle, M.; Pauli, J.; Spieles, M.; Resch-Genger, U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 2013, 8, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.J.; Gale, P.A. Changing and challenging times for service crystallography. Chem. Sci. 2012, 3, 683–689. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Campos, E.; Moura, T.F.; Oliva, A.; Leandro, P.; Soveral, G. Lack of Aquaporin 3 in bovine erythrocyte membranes correlates with low glycerol permeation. Biochem. Biophys. Res. Commun. 2011, 408, 477–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AQP3 Inhibition | EC50 [μM] 1 | |||
|---|---|---|---|---|---|
| IC50 [μM] 1 | SKOV-3 | A375 | MCF7 | A549 | |
| Auphen | 0.80 ± 0.08 | 7.00 ± 2.00 | 1.7 ± 0.3 | 3.00 ± 0.05 | 1.07 ± 0.09 |
| C1 | 1.018 ± 0.137 | >80 | >80 | >80 | >80 |
| C2 | 0.881 ± 0.015 | >80 | >80 | >80 | >80 |
| C3 | 1.825 ± 0.017 (n = 2) | 41 ± 13 | 23 ± 1 | 40 ± 4 | 57 ± 2 |
| C4 | 0.85 ± 0.21 | 56 ± 12 | 69 ± 3 | 63 ± 1 | 81 ± 9 |
| C5 | 0.80 ± 0.10 | >50 | >50 | 25 (n = 1) | >50) |
| C6 | 0.69 ± 0.06 | >50 | 34 (n = 1) | 38 (n = 1) | 47 (n = 2) |
| C7 | n.d. | n.d. | n.d. | n.d. | n.d. |
| C8 | n.d. | n.d. | n.d. | n.d. | n.d. |
| C9 | 0.72 ± 0.05 | >50 | >50 | >50 | >50 |
| C10 | >50 | 17 ± 7 | 5 ± 2 | 12 ± 1 | >50 |
| C11 | 0.82 ± 0.13 (n = 2) | 33 ± 5 | 12 ± 2 | 29 ± 8 | >50 |
| C12 | n.d. | 41 ±13 | 13 ± 2 | 17 ± 3 | 45 ± 3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aikman, B.; Wenzel, M.N.; Mósca, A.F.; De Almeida, A.; Klooster, W.T.; Coles, S.J.; Soveral, G.; Casini, A. Gold(III) Pyridine-Benzimidazole Complexes as Aquaglyceroporin Inhibitors and Antiproliferative Agents. Inorganics 2018, 6, 123. https://doi.org/10.3390/inorganics6040123
Aikman B, Wenzel MN, Mósca AF, De Almeida A, Klooster WT, Coles SJ, Soveral G, Casini A. Gold(III) Pyridine-Benzimidazole Complexes as Aquaglyceroporin Inhibitors and Antiproliferative Agents. Inorganics. 2018; 6(4):123. https://doi.org/10.3390/inorganics6040123
Chicago/Turabian StyleAikman, Brech, Margot N. Wenzel, Andreia F. Mósca, Andreia De Almeida, Wim T. Klooster, Simon J. Coles, Graça Soveral, and Angela Casini. 2018. "Gold(III) Pyridine-Benzimidazole Complexes as Aquaglyceroporin Inhibitors and Antiproliferative Agents" Inorganics 6, no. 4: 123. https://doi.org/10.3390/inorganics6040123
APA StyleAikman, B., Wenzel, M. N., Mósca, A. F., De Almeida, A., Klooster, W. T., Coles, S. J., Soveral, G., & Casini, A. (2018). Gold(III) Pyridine-Benzimidazole Complexes as Aquaglyceroporin Inhibitors and Antiproliferative Agents. Inorganics, 6(4), 123. https://doi.org/10.3390/inorganics6040123