Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis


Abstract
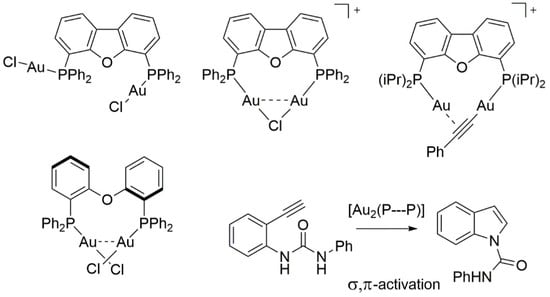
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
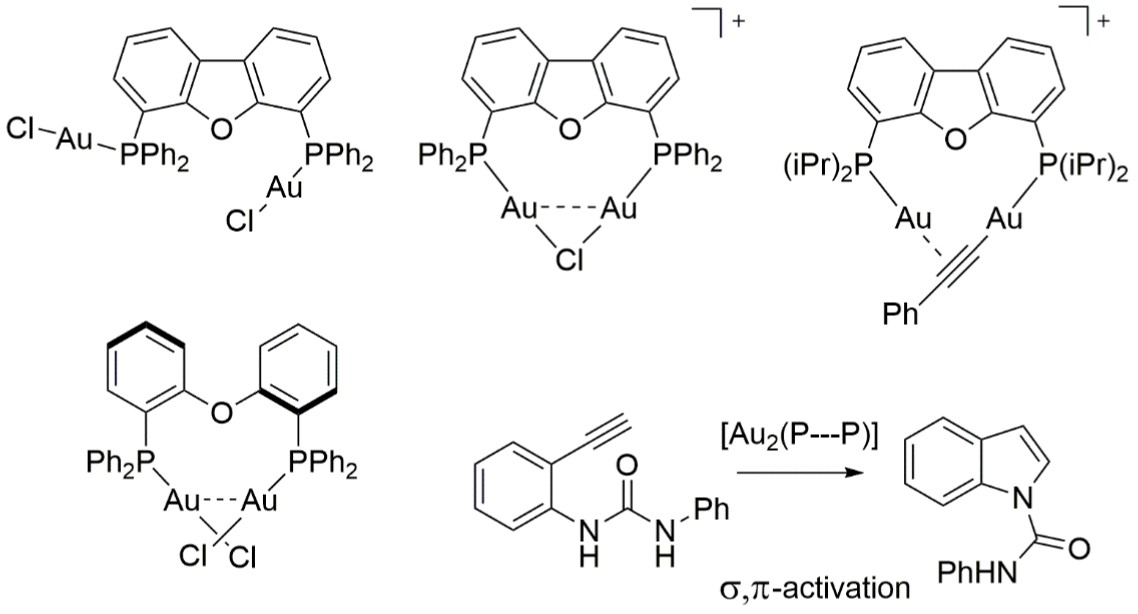
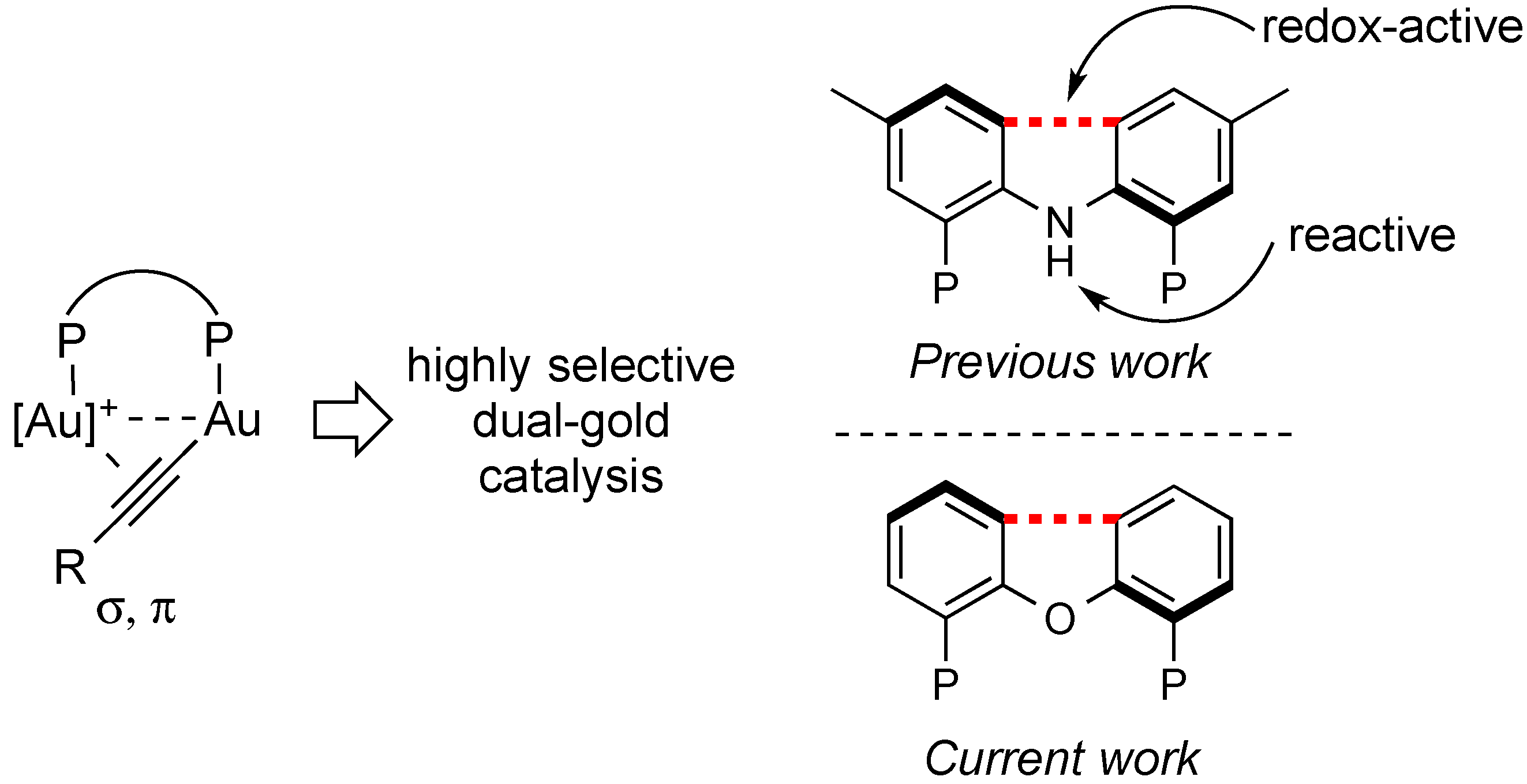
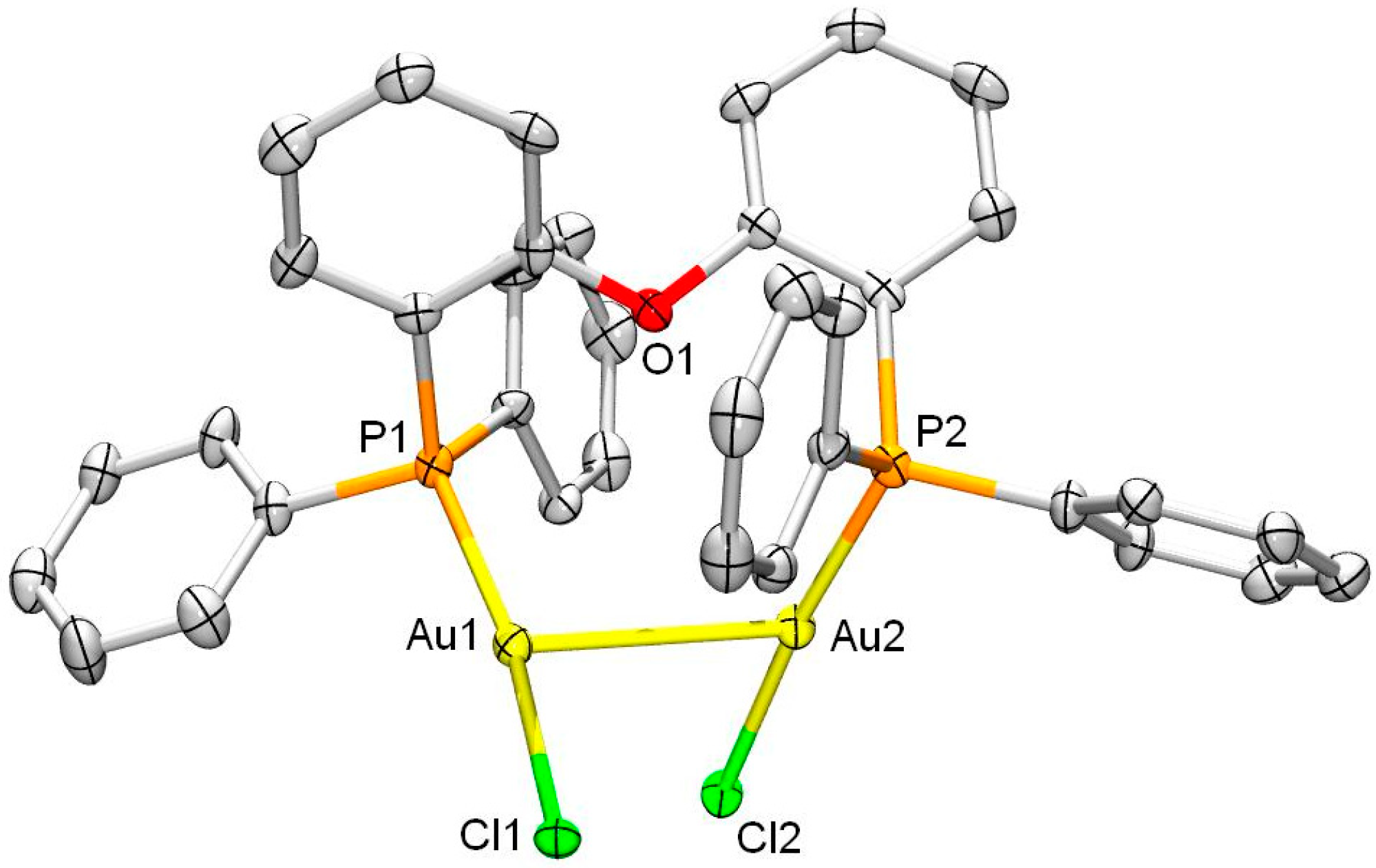
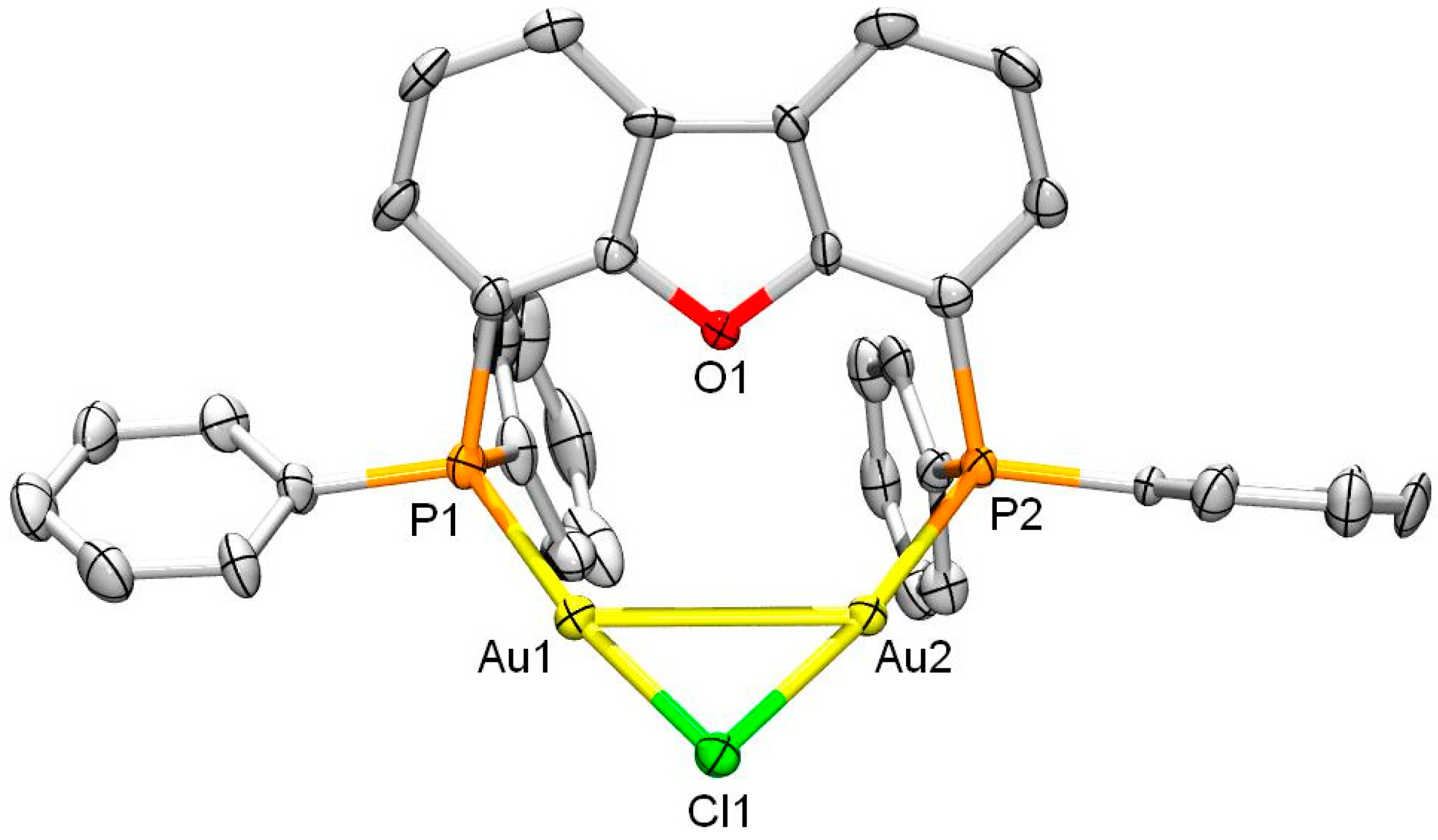
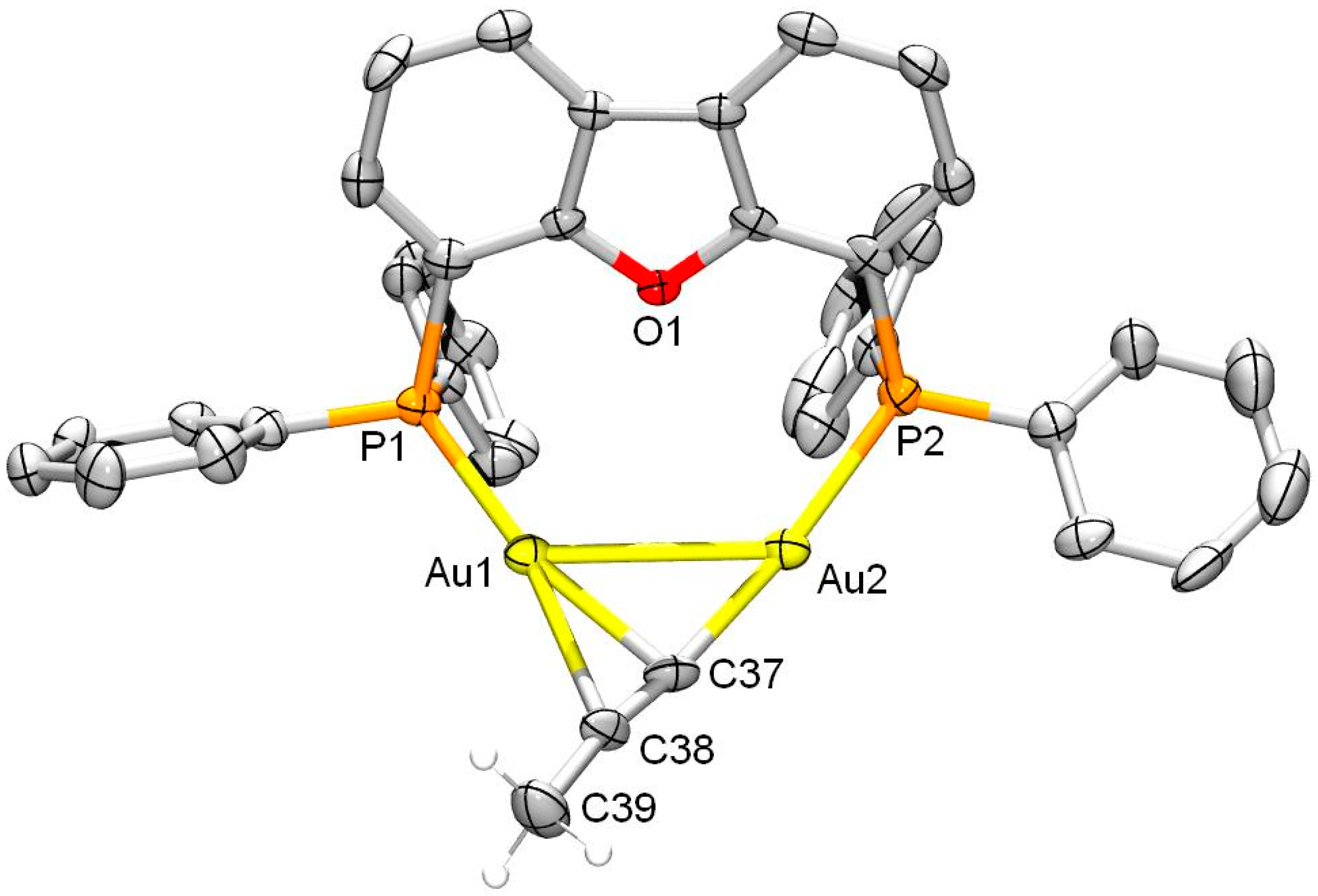
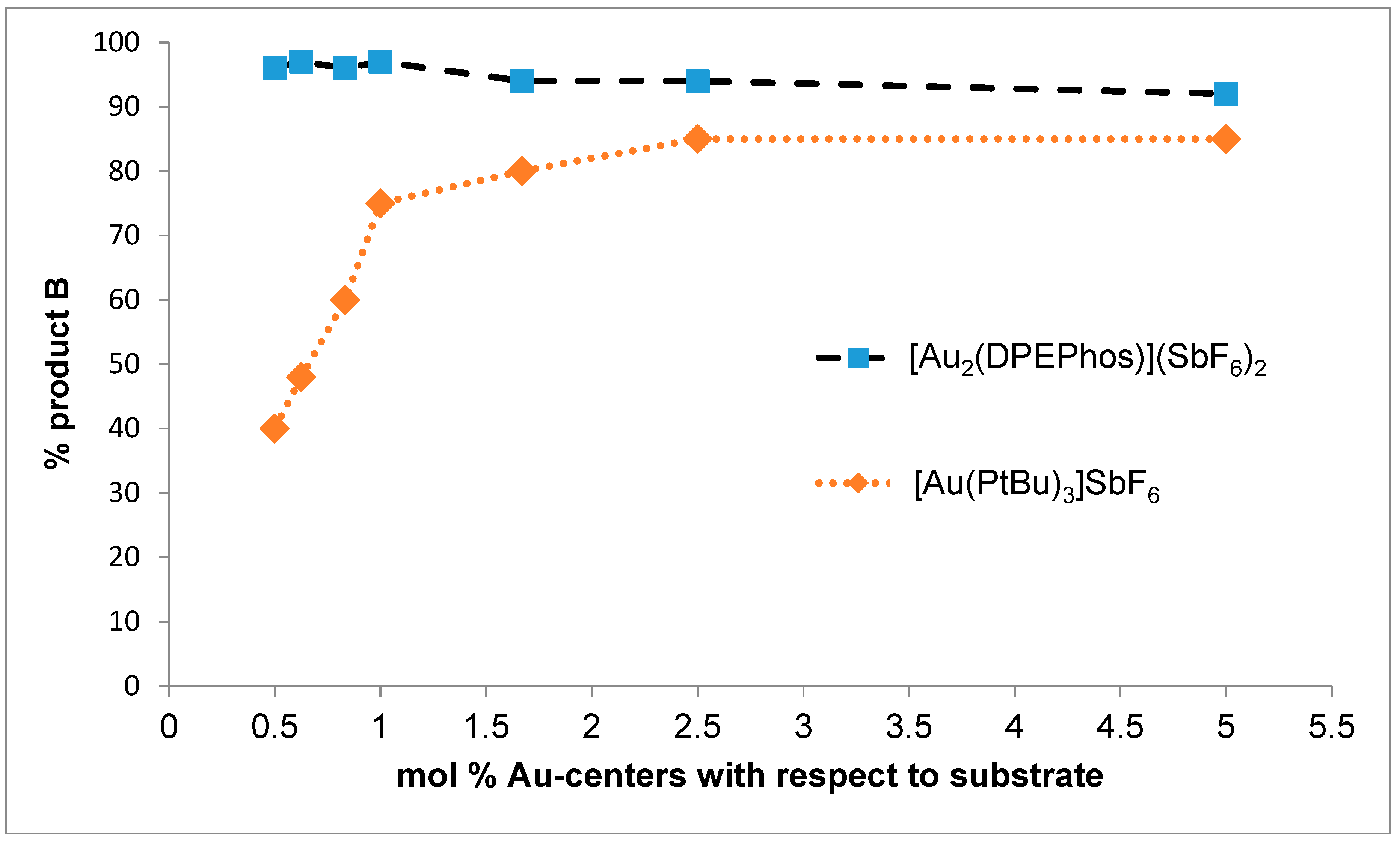
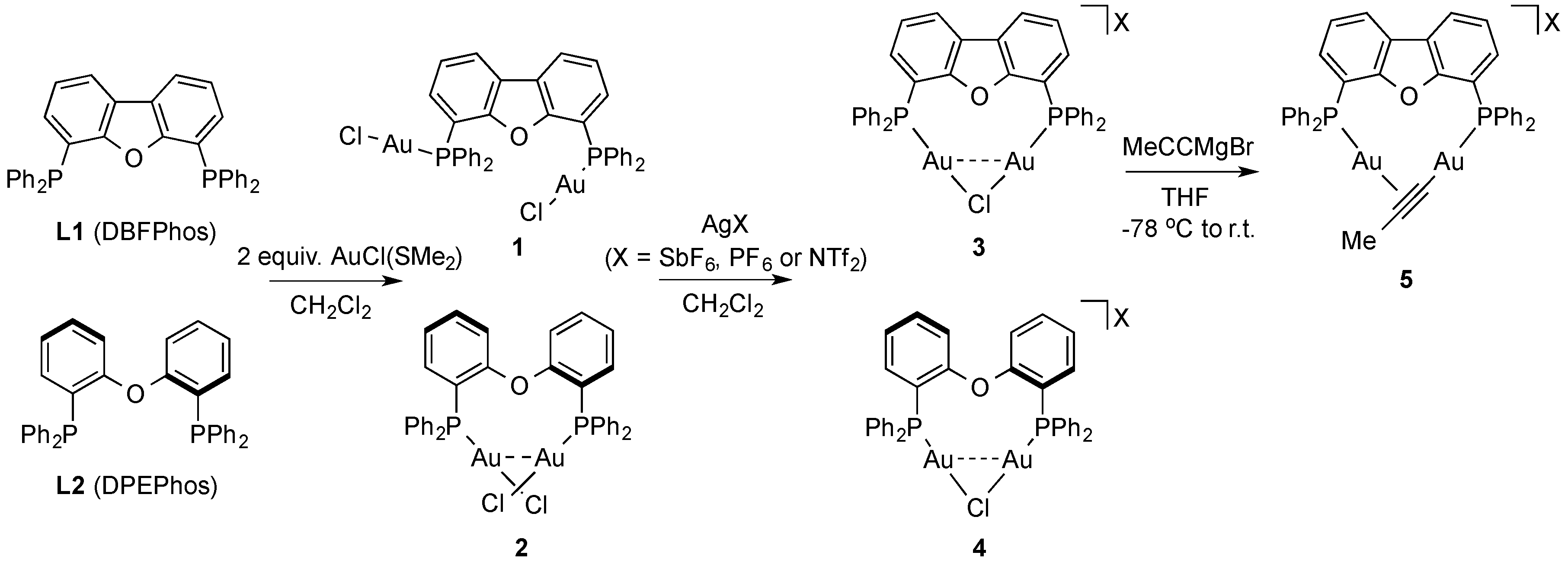
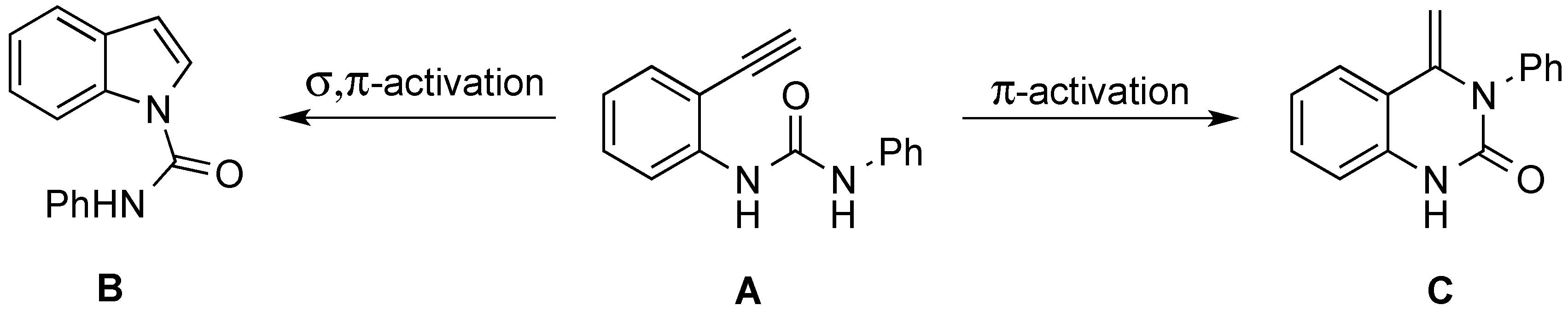
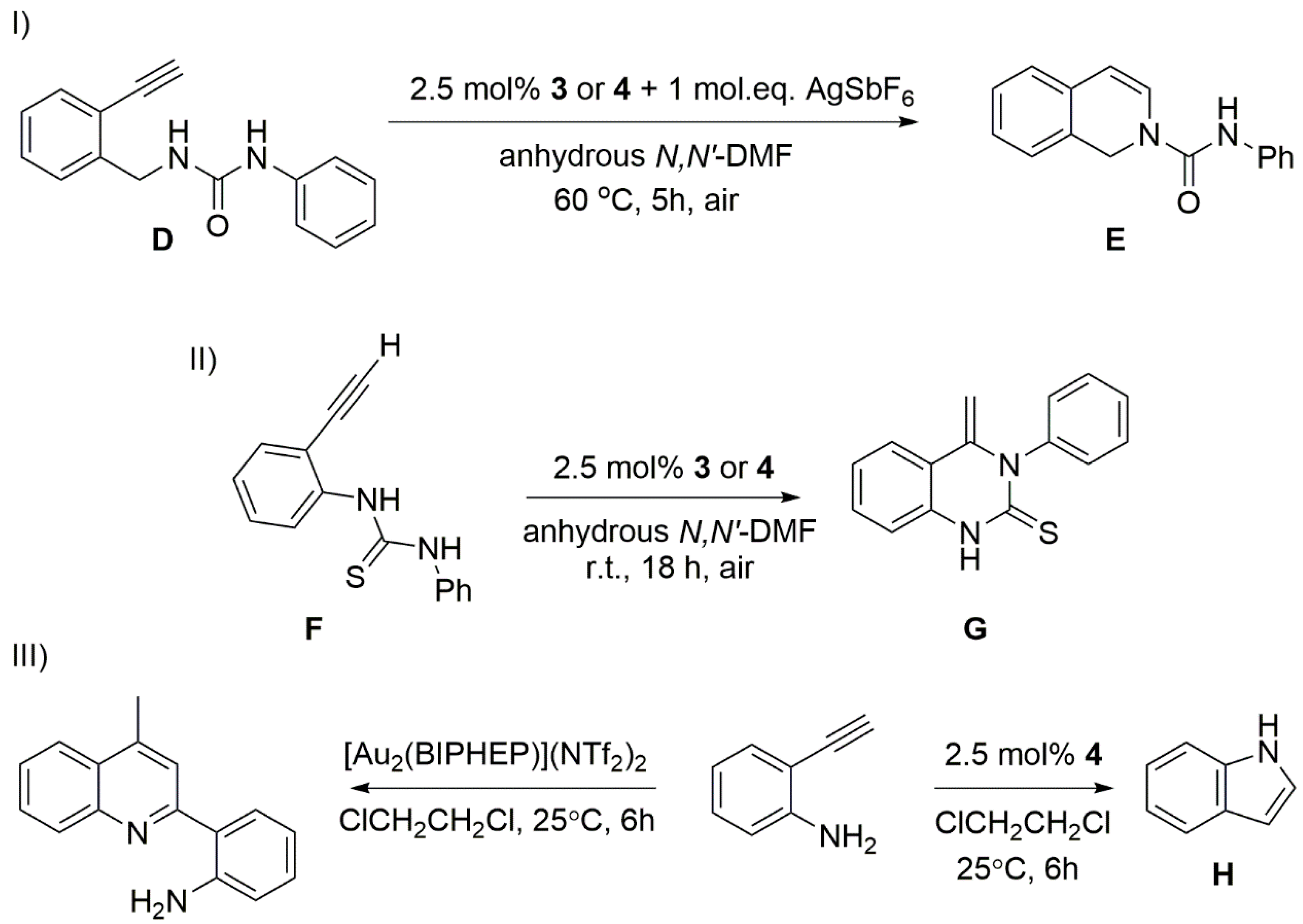
2. Results and Discussion
3. Materials and Methods
3.1. General Comments
3.2. Syntheses of New Complexes
3.3. General Information on Catalytic Studies
3.4. Single Crystal X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hashmi, A.S.K. Dual-Gold Catalysis. Acc. Chem. Res. 2014, 47, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Chiarucci, M.; Bandini, M. New developments in gold-catalyzed manipulation of inactivated alkenes. Beilstein J. Org. Chem. 2013, 9, 2586–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranieri, B.; Escofet, I.; Echavarren, A.M. Anatomy of gold catalysts: Facts and myths. Org. Biomol. Chem. 2015, 13, 7103–7118. [Google Scholar] [CrossRef] [PubMed]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Shi, M. Divergent Synthesis of Carbo- and Heterocycles via Gold-Catalyzed Reactions. ACS Catal. 2016, 6, 2515–2524. [Google Scholar] [CrossRef]
- Pflästerer, D.; Hashmi, A.S.K. Gold catalysis in total synthesis—Recent achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. [Google Scholar] [CrossRef] [PubMed]
- Jans, A.C.H.; Caumes, X.; Reek, J.N.H. Gold Catalysis in (Supra)Molecular Cages to Control Reactivity and Selectivity. ChemCatChem 2019, 11, 287–297. [Google Scholar] [CrossRef]
- Cheong, P.H.-Y.; Morganelli, P.; Luzung, M.R.; Houk, K.N.; Toste, F.D. Gold-Catalyzed Cycloisomerization of 1,5-Allenynes via Dual Activation of an Ene Reaction. J. Am. Chem. Soc. 2008, 130, 4517–4526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grirrane, A.; Garcia, H.; Corma, A.; Álvarez, E. Intermolecular [2 + 2] Cycloaddition of Alkyne-Alkene Catalyzed by Au(I) Complexes. What Are the Catalytic Sites Involved? ACS Catal. 2011, 1, 1647–1653. [Google Scholar] [CrossRef] [Green Version]
- Hashmi, A.S.K.; Braun, I.; Nösel, P.; Schädlich, J.; Wieteck, M.; Rudolph, M.; Rominger, F. Simple Gold-Catalyzed Synthesis of Benzofulvenes—gem-Diaurated Species as “Instant Dual-Activation” Precatalysts. Angew. Chem. Int. Ed. 2012, 51, 4456–4460. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Braun, I.; Rudolph, M.; Rominger, F. The Role of Gold Acetylides as a Selectivity Trigger and the Importance of gem-Diaurated Species in the Gold-Catalyzed Hydroarylating-Aromatization of Arene-Diynes. Organometallics 2012, 31, 644–661. [Google Scholar] [CrossRef]
- Ye, L.; Wang, Y.; Aue, D.H.; Zhang, L. Experimental and Computational Evidence for Gold Vinylidenes: Generation from Terminal Alkynes via a Bifurcation Pathway and Facile C–H Insertions. J. Am. Chem. Soc. 2012, 134, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Oonishi, Y.; Gómez-Suárez, A.; Martin, A.R.; Nolan, S.P. Hydrophenoxylation of Alkynes by Cooperative Gold Catalysis. Angew. Chem. Int. Ed. 2013, 52, 9767–9771. [Google Scholar] [CrossRef] [PubMed]
- Højer Larsen, M.; Houk, K.N.; Hashmi, A.S.K. Dual Gold Catalysis: Stepwise Catalyst Transfer via Dinuclear Clusters. J. Am. Chem. Soc. 2015, 137, 10668–10676. [Google Scholar] [CrossRef] [PubMed]
- Tkatchouk, E.; Mankad, N.P.; Benitez, D.; Goddard, W.A.; Toste, F.D. Two Metals Are Better Than One in the Gold Catalyzed Oxidative Heteroarylation of Alkenes. J. Am. Chem. Soc. 2011, 133, 14293–14300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadie, M.-A.; Trivelli, X.; Medina, F.; Capet, F.; Roussel, P.; Agbossou-Niedercorn, F.; Michon, C. Asymmetric Intramolecular Hydroamination of Alkenes in Mild and Wet Conditions—Structure and Reactivity of Cationic Binuclear Gold(I) Catalysts. ChemCatChem 2014, 6, 2235–2239. [Google Scholar] [CrossRef]
- Serrano-Becerra, J.M.; Maier, A.F.G.; González-Gallardo, S.; Moos, E.; Kaub, C.; Gaffga, M.; Niedner-Schatteburg, G.; Roesky, P.W.; Breher, F.; Paradies, J. Mono- vs. Dinuclear Gold-Catalyzed Intermolecular Hydroamidation. Eur. J. Org. Chem. 2014, 4515–4522. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Lauterbach, T.; Nösel, P.; Vilhelmsen, M.H.; Rudolph, M.; Rominger, F. Dual Gold Catalysis: σ,π-Propyne Acetylide and Hydroxyl-Bridged Digold Complexes as Easy-To-Prepare and Easy-To-Handle Precatalysts. Chem. Eur. J. 2012, 19, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Vreeken, V.; Broere, D.L.J.; Jans, A.C.H.; Lankelma, M.; Reek, J.N.H.; Siegler, M.A.; van der Vlugt, J.I. Well-Defined Dinuclear Gold Complexes for Preorganization-Induced Selective Dual Gold Catalysis. Angew. Chem. Int. Ed. 2016, 55, 10042–10046. [Google Scholar] [CrossRef] [PubMed]
- Vreeken, V.; Siegler, M.A.; van der Vlugt, J.I. Controlled Interconversion of a Dinuclear Au Species Supported by a Redox-Active Bridging PNP Ligand Facilitates Ligand-to-Gold Electron Transfer. Chem. Eur. J. 2017, 23, 5585–5594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamer, P.C.J.; van Leeuwen, P.W.N.M. Phosphorus(III) Li Gands in Homogeneous Catalysis: Design and Synthesis; Wiley: Chichester, UK, 2012. [Google Scholar]
- Van Leeuwen, P.W.N.M.; Kamer, P.C.J. Featuring Xantphos. Catal. Sci. Technol. 2018, 8, 26–113. [Google Scholar] [CrossRef]
- Kamer, P.C.J.; van Leeuwen, P.W.N.M.; Reek, J.N.H. Wide Bite Angle Diphosphines: Xantphos Ligands in Transition Metal Complexes and Catalysis. Acc. Chem. Res. 2001, 34, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, M.; van der Burgt, Y.E.M.; Kamer, P.C.J.; van Leeuwen, P.W.N.M.; Goubitz, K.; Fraanje, J. New Diphosphine Ligands Based on Heterocyclic Aromatics Inducing Very High Regioselectivity in Rhodium-Catalyzed Hydroformylation: Effect of the Bite Angle. Organometallics 1995, 14, 3081–3089. [Google Scholar] [Green Version]
- Van der Vlugt, J.I.; van Duren, R.; Batema, G.D.; den Heeten, R.; Meetsma, A.; Fraanje, J.; Goubitz, K.; Kamer, P.C.J.; van Leeuwen, P.W.N.M.; Vogt, D. Platinum Complexes of Rigid Bidentate Phosphine Ligands in the Hydroformylation of 1-Octene. Organometallics 2005, 24, 5377–5382. [Google Scholar] [CrossRef]
- Van Duren, R.; van der Vlugt, J.I.; Mills, A.M.; Spek, A.L.; Vogt, D. Platinum-catalyzed hydroformylation of terminal and internal octenes. Dalton Trans. 2007, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Czauderna, C.F.; Cordes, D.B.; Slawin, A.M.Z.; Müller, C.; van der Vlugt, J.I.; Vogt, D.; Kamer, P.C.J. Synthesis and reactivity of chiral wide bite angle hybrid diphosphorus ligands. Eur. J. Inorg. Chem. 2014, 1797–1810. [Google Scholar] [CrossRef]
- Czauderna, C.F.; Jarvis, A.G.; Heutz, F.J.L.; Cordes, D.B.; Slawin, A.M.Z.; van der Vlugt, J.I.; Kamer, P.C.J. Chiral Wide-Bite-Angle Diphosphine Ligands: Synthesis, Coordination Chemistry, and Application in Pd-Catalyzed Allylic Alkylation. Organometallics 2015, 34, 1608–1618. [Google Scholar] [CrossRef]
- Czauderna, C.F.; Slawin, A.M.Z.; Cordes, D.B.; van der Vlugt, J.I.; Kamer, P.C.J. P-stereogenic wide bite angle diphosphine ligands. Tetrahedron 2019, 75, 47–56. [Google Scholar] [CrossRef]
- De la Riva, H.; Nieuwhuyzen, M.; Mendicute Fierro, C.; Raithby, P.R.; Male, L.; Lagunas, M.C. A New Type of Luminescent Alkynyl Au4Cu2 Cluster. Inorg. Chem. 2006, 45, 1418–1420. [Google Scholar] [CrossRef] [PubMed]
- Pintado-Alba, A.; de la Riva, H.; Nieuwhuyzen, M.; Bautista, D.; Raithby, P.R.; Sparkes, H.A.; Teat, S.J.; López-de-Luzuriaga, J.M.; Lagunas, M.C. Effects of diphosphine structure on aurophilicity and luminescence in Au(I) complexes. Dalton Trans. 2004, 3459–3467. [Google Scholar] [CrossRef] [PubMed]
- Partyka, D.V.; Updegraff III, J.B.; Zeller, M.; Hunter, A.D.; Gray, T.G. Gold(I) halide complexes of bis(diphenylphosphine)diphenyl ether ligands: A balance of ligand strain and non-covalent interactions. Dalton Trans. 2010, 39, 5388–5397. [Google Scholar] [CrossRef] [PubMed]
- Glebko, N.; Dau, T.M.; Melnikov, A.S.; Grachova, E.V.; Solovyev, I.V.; Belyaev, A.; Karttunen, A.J.; Koshevoy, I.O. Luminescence Thermochromism of Gold(I) Phosphane–Iodide Complexes: A Rule or an Exception? Chem. Eur. J. 2018, 24, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Jobbágy, C.; Baranai, P.; Marsi, G.; Rácz, B.; Li, L.; Naumov, P.; Deák, A. Novel gold(I) diphosphine-based dimers with aurophilicity triggered multistimuli light-emitting properties. J. Mater. Chem. C 2016, 4, 10253–10264. [Google Scholar] [CrossRef]
- Jobbágy, C.; Baranai, P.; Szabó, P.; Holczbauer, T.; Rácz, B.; Li, L.; Naumov, P.; Deák, A. Unexpected formation of a fused double cycle trinuclear gold(I) complex supported by ortho-phenyl metallated aryl-diphosphine ligands and strong aurophilic interactions. Dalton Trans. 2016, 45, 12569–12575. [Google Scholar] [CrossRef] [PubMed]
- Baranai, P.; Marsi, G.; Hamza, A.; Jobbágy, C.; Deák, A. Structural characterization of dinuclear gold(I) diphosphine complexes with anion-triggered luminescence. Struct. Chem. 2015, 26, 1377–1387. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Saito, T.; Miyahara, T.; Zhong, C.; Sawamura, M. Gold(I) Hydride Intermediate in Catalysis: Dehydrogenative Alcohol Silylation Catalyzed by Gold(I) Complex. Organometallics 2009, 28, 4829–4840. [Google Scholar] [CrossRef]
- Deák, A.; Megyes, T.; Tárkányi, G.; Király, P.; Biczók, L.; Pálinkás, G.; Stang, P.J. Synthesis and Solution- and Solid-State Characterization of Gold(I) Rings with Short Au···Au Interactions. Spontaneous Resolution of a Gold(I) Complex. J. Am. Chem. Soc. 2006, 128, 12668–12670. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Schier, A. A briefing on aurophilicity. Chem. Soc. Rev. 2008, 37, 1931–1951. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.T.; England, K.R.; Ghiassi, K.B.; Semma, F.Z.; Olmstead, M.M.; Balch, A.L. Steric effects and aurophilic interactions in crystals of Au2(μ-1,2-bis(diphenylphosphino)ethane)X2 and Au2(μ-1,2-bis(dicyclohexylphosphino)ethane)X2 (X = Cl, Br, I). Polyhedron 2016, 117, 535–541. [Google Scholar] [CrossRef]
- Mir, M.H.; Ong, J.X.; Kole, G.K.; Tan, G.K.; McGlinchey, M.J.; Wu, Y.; Vittal, J.J. Photoreactive gold(I) macrocycles with diphosphine and trans,trans-muconate ligands. Chem. Commun. 2011, 47, 11633–11635. [Google Scholar] [CrossRef] [PubMed]
- Grirrane, A.; Álvarez, E.; García, H.; Corma, A. Deactivation of Cationic CuI and AuI Catalysts for A3 Coupling by CH2Cl2: Mechanistic Implications of the Formation of Neutral CuI and AuI Chlorides. Angew. Chem. Int. Ed. 2014, 53, 7253–7258. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.; Dodson, T.; Tirfoin, R.; Bates, J.I.; Aldridge, S. Expanded-Ring N-Heterocyclic Carbenes for the Stabilization of Highly Electrophilic Gold(I) Cations. Chem. Eur. J. 2014, 20, 16721–16731. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Day, C.S.; Zhang, L.; Hauser, K.J.; Jones, A.C. A Unique Au–Ag–Au Triangular Motif in a Trimetallic Halonium Dication: Silver Incorporation in a Gold(I) Catalyst. Chem. Eur. J. 2013, 19, 12264–12271. [Google Scholar] [CrossRef] [PubMed]
- Homs, A.; Escofet, I.; Echavarren, A.M. On the Silver Effect and the Formation of Chloride-Bridged Digold Complexes. Org. Lett. 2013, 15, 5782–5785. [Google Scholar] [CrossRef] [PubMed]
- Yam, V.W.W.; Chan, C.-L.; Cheung, K.-K. Synthesis and photophysics of dinuclear gold(I) thiolates of bis(diphenylphosphino)-alkyl- and -aryl-amines. Crystal structure of [Au2{Ph2PN(C6H11)PPh2}(SC6H4F-p)2]. J. Chem. Soc. Dalton Trans. 1996, 4019–4022. [Google Scholar] [CrossRef]
- Jones, P.G.; Sheldrick, G.M.; Uson, R.; Lapuna, A. μ-Chloro-bis(triphenylphosphine) digold(I) perchlorate dichloromethane solvate. Acta Cryst. B 1980, 36, 1486. [Google Scholar] [CrossRef]
- Hamel, A.; Mitzel, N.W.; Schmidbaur, H. Metallophilicity: The Dimerization of Bis[(triphenylphosphine)gold(I)]chloronium Cations. J. Am. Chem. Soc. 2001, 123, 5106–5107. [Google Scholar] [CrossRef]
- Gimeno, A.; Cuenca, A.B.; Suárez-Pantiga, S.; Ramírez de Arellano, C.; Medio-Simón, M.; Asensio, G. Competitive Gold-Activation Modes in Terminal Alkynes: An Experimental and Mechanistic Study. Chem. Eur. J. 2014, 20, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Gramage-Doria, R.; Hessels, J.; Leenders, S.H.A.M.; Tröppner, O.; Dürr, M.; Ivanović-Burmazović, I.; Reek, J.N.H. Gold(I) Catalysis at Extreme Concentrations Inside Self-Assembled Nanospheres. Angew. Chem. Int. Ed. 2014, 53, 13380–13384. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Q.; Gonell, S.; Leenders, S.H.A.M.; Tröppner, O.; Dürr, M.; Ivanović-Burmazović, I.; Reek, J.N.H. Self-assembled nanospheres with multiple endohedral binding sites pre-organize catalysts and substrates for highly efficient reactions. Nat. Chem. 2016, 8, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Wang, J.; Zhang, X.; Zhou, Y.; Ding, X.; Feng, E.; Sun, H.; Liu, G.; Jiang, H.; Liu, H. Gold-catalyzed intramolecular hydroamination of terminal alkynes in aqueous media: Efficient and regioselective synthesis of indole-1-carboxamides. Green Chem. 2009, 11, 1201–1208. [Google Scholar] [CrossRef]
- Jiang, Y.; Wei, Y.; Tang, X.Y.; Shi, M. Gold(I)-Catalyzed Selective Heterocyclization of Propargylic Thioureas: Mechanistic Study of Competitive Gold-Activation Mode. Gold(I)-Catalyzed Selective Heterocyclization of Propargylic Thioureas: Mechanistic Study of Competitive Gold-Activation Mode. Chem. Eur. J. 2015, 21, 7675–7681. [Google Scholar] [CrossRef] [PubMed]
- Praveen, C.; Perumal, P. Revisiting the Gold-Catalyzed Dimerization of 2-Ethynylanilines: A Room-Temperature and Silver-Free Protocol for the Synthesis of Multifunctional Quinolines. Synthesis 2016, 48, 855–864. [Google Scholar] [CrossRef]
- Varela-Fernández, A.; Varela, J.A.; Saá, C. Formation of Indoles, Dihydroisoquinolines, and Dihydroquinolines by Ruthenium-Catalyzed Heterocyclizations. Synthesis 2012, 44, 3285–3295. [Google Scholar]
- Bruker. APEX2 Software; Bruker: Madison, WI, USA, 2014. [Google Scholar]
- Sheldrick, G.M. SADABS; Universität Göttingen: Lower Saxony, Germany, 2008. [Google Scholar]
- Sheldrick, G.M. SHELXL2013; University of Göttingen: Lower Saxony, Germany, 2013. [Google Scholar]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lankelma, M.; Vreeken, V.; Siegler, M.A.; van der Vlugt, J.I. Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis. Inorganics 2019, 7, 28. https://doi.org/10.3390/inorganics7030028
Lankelma M, Vreeken V, Siegler MA, van der Vlugt JI. Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis. Inorganics. 2019; 7(3):28. https://doi.org/10.3390/inorganics7030028
Chicago/Turabian StyleLankelma, Marianne, Vincent Vreeken, Maxime A. Siegler, and Jarl Ivar van der Vlugt. 2019. "Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis" Inorganics 7, no. 3: 28. https://doi.org/10.3390/inorganics7030028
APA StyleLankelma, M., Vreeken, V., Siegler, M. A., & van der Vlugt, J. I. (2019). Dinuclear Gold Complexes Supported by Wide Bite Angle Diphosphines for Preorganization-Induced Selective Dual-Gold Catalysis. Inorganics, 7(3), 28. https://doi.org/10.3390/inorganics7030028