Synthesis, Structures and Chemistry of the Metallaboranes of Group 4–9 with M2B5 Core Having a Cross Cluster M–M Bond

 , , ,
, , , 
Abstract
:
1. Introduction
2. Results and Discussion
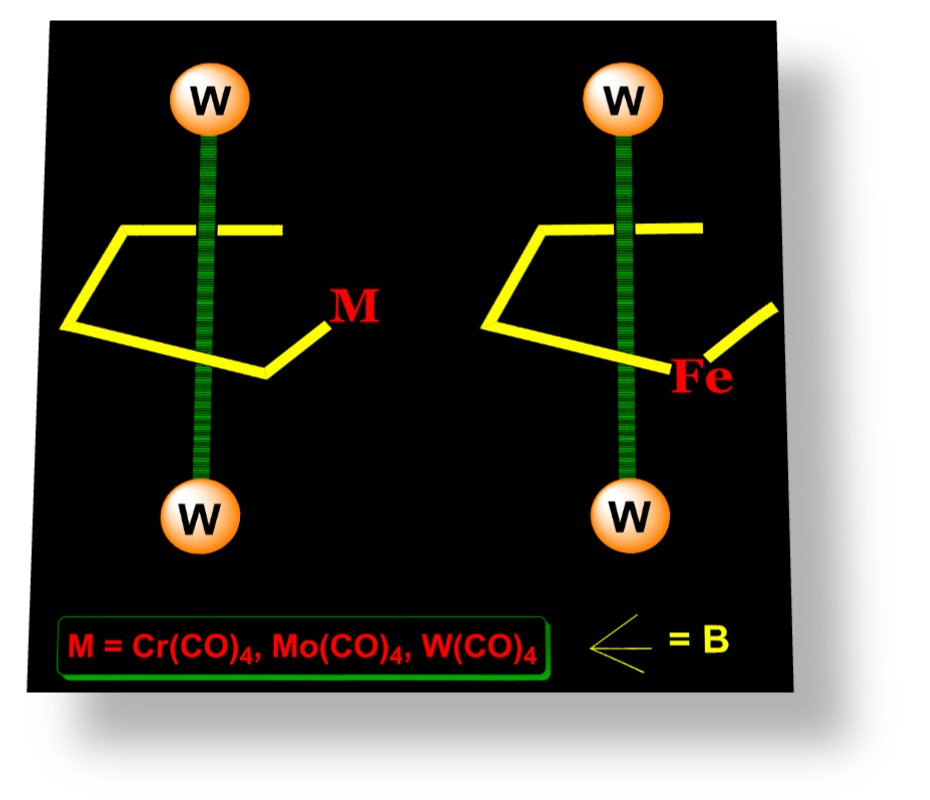
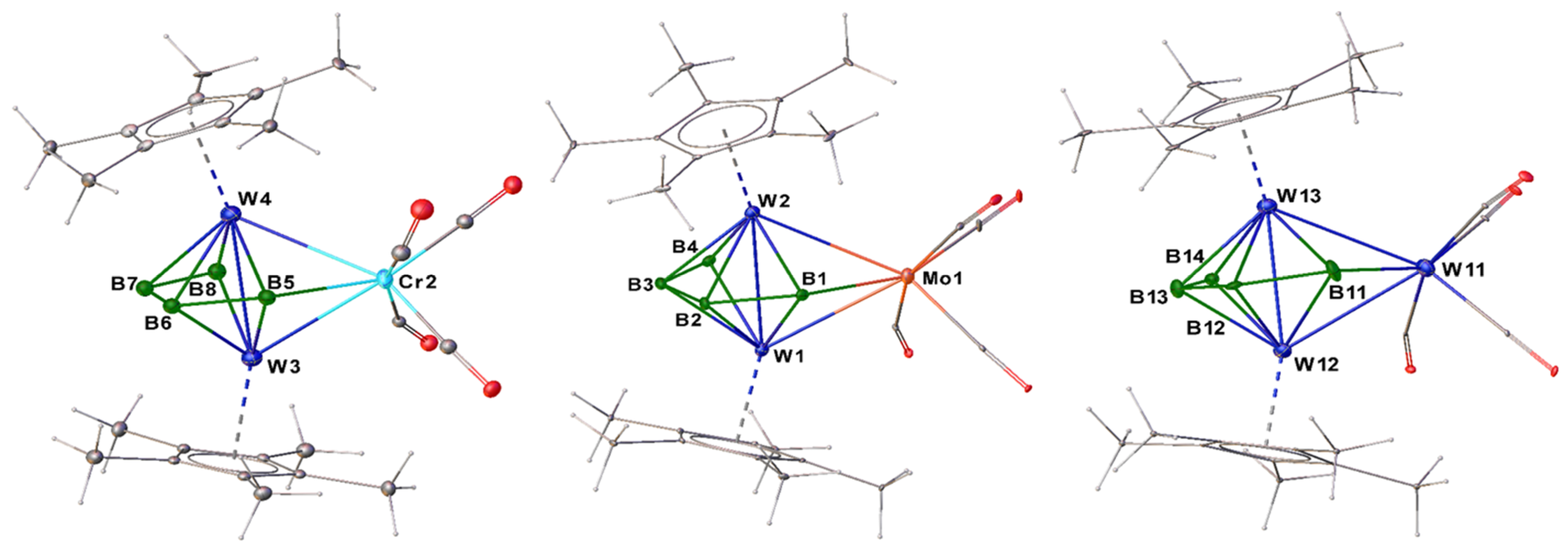
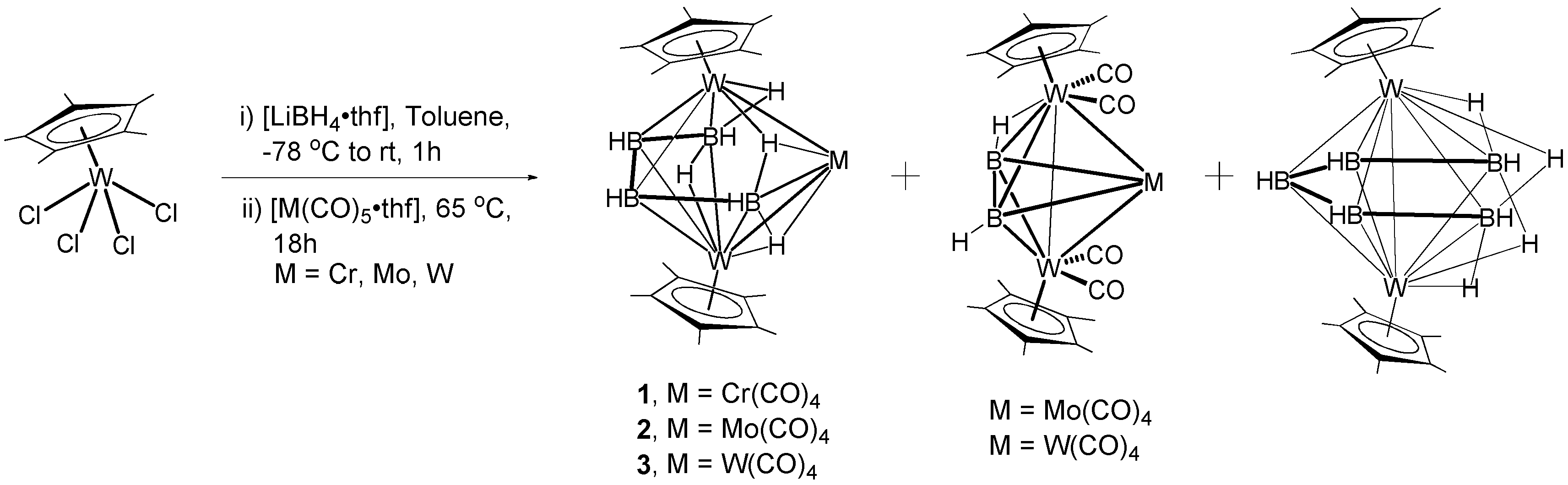
2.1. Synthesis and Characterization of Tungstaboranes [(Cp*W)2B4H8M(CO)4], 1–3 (1: M = Cr; 2: M = Mo; 3: M = W)
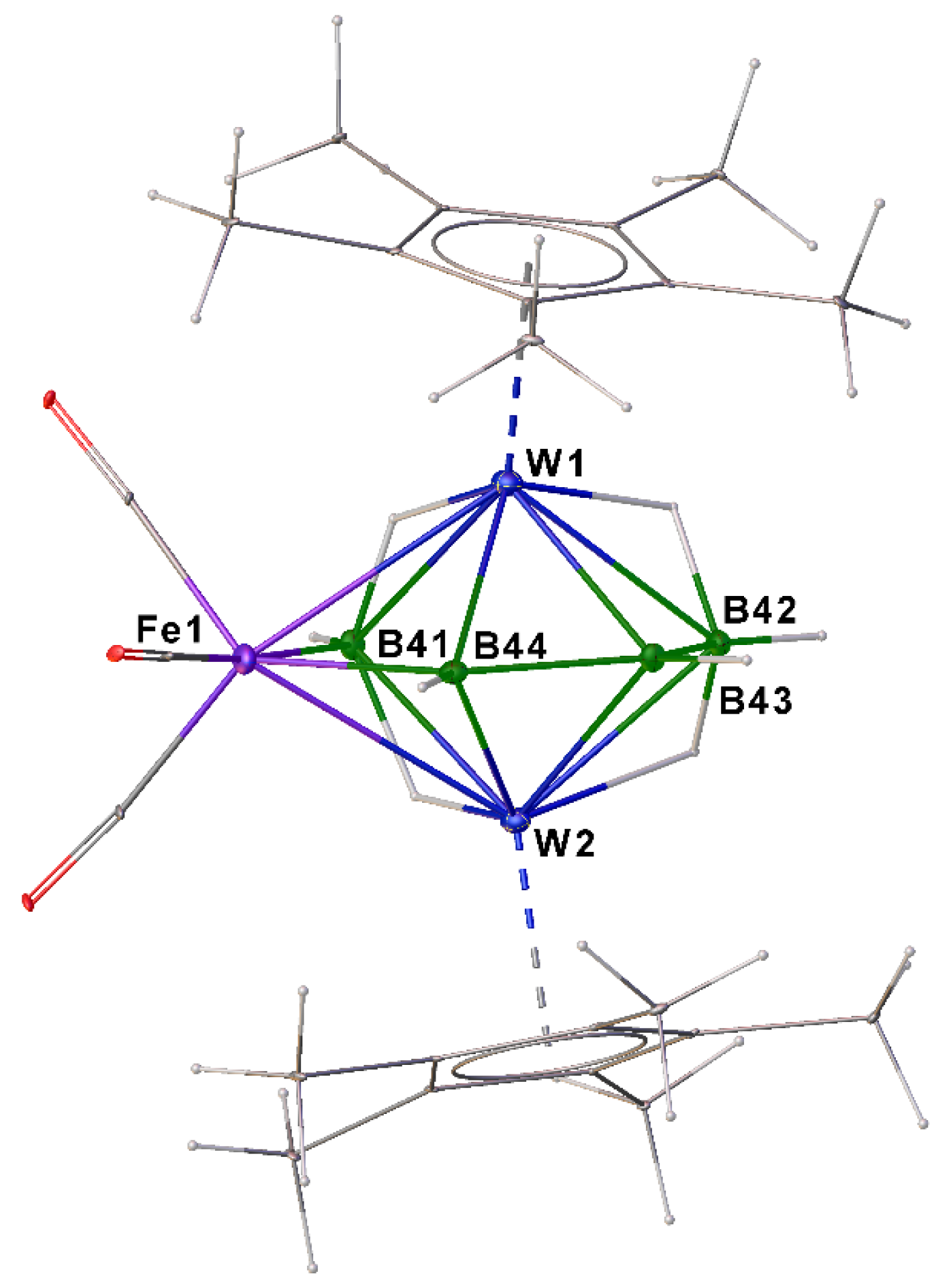
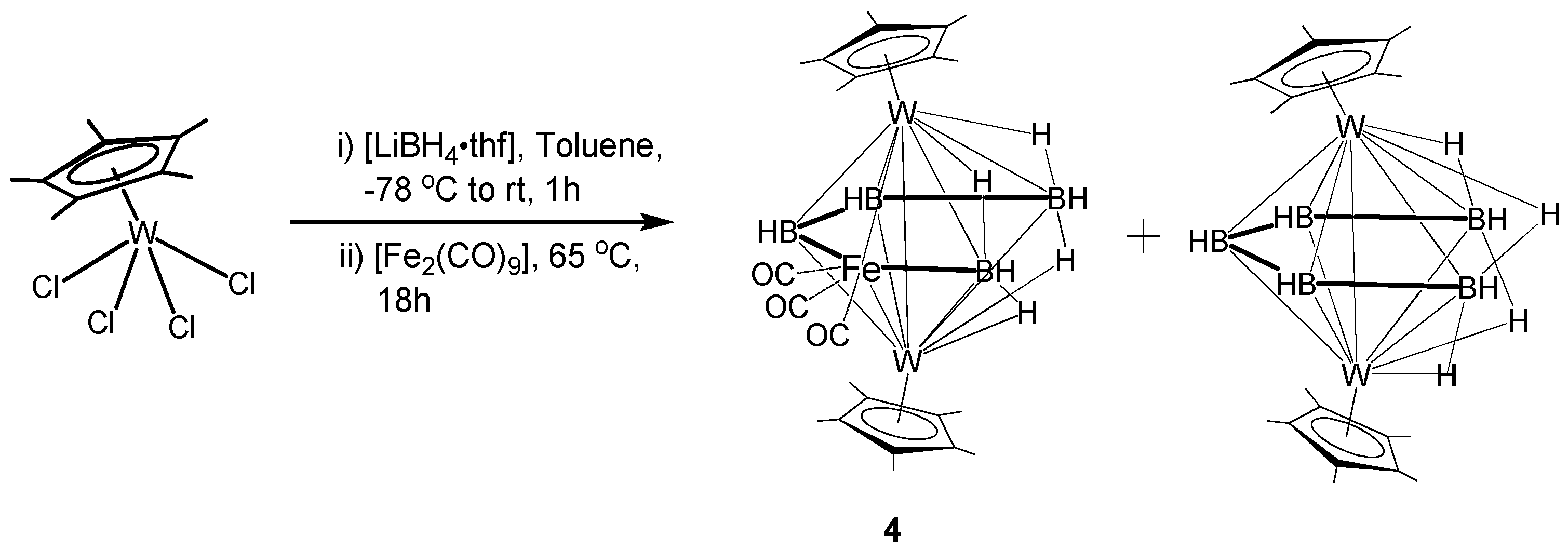
2.2. Synthesis and Characterization of Mixed Metal Tungstaborane [(Cp*W)2B4H8Fe(CO)3], 4
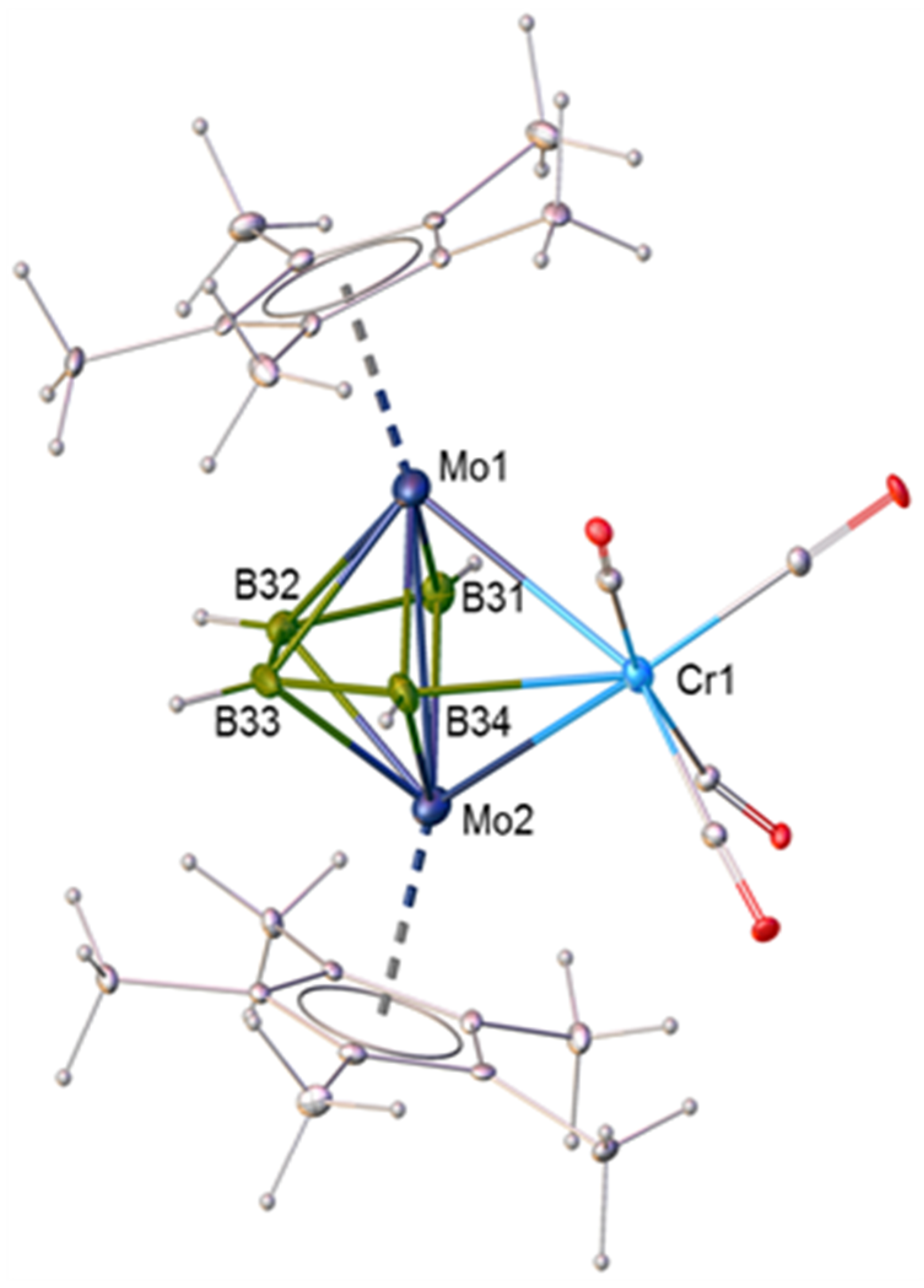
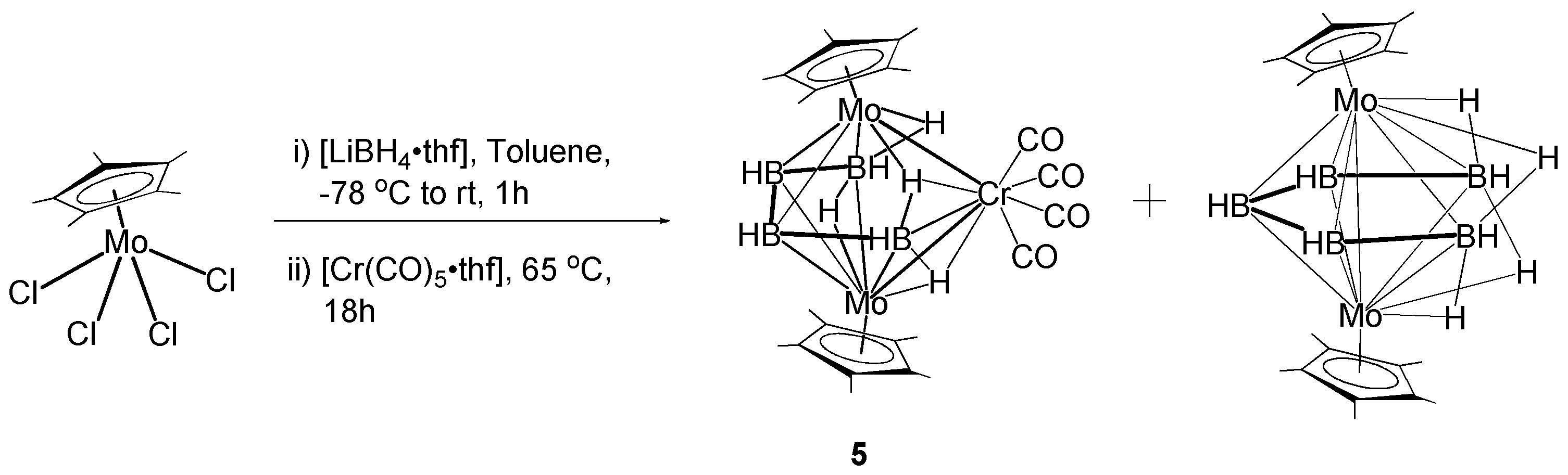
2.3. Synthesis and Characterization of the Molybdaborane [(Cp*Mo)2B4H8Cr(CO)4], 5
3. Materials and Methods
3.1. General Procedures and Instrumentation
3.2. X-ray Structure Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fehlner, T.P.; Halet, J.-F.; Saillard, J.-Y. Molecular Clusters: A Bridge to Solid-State Chemistry; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Greenwood, N.N.; Ward, I.M. Metalloboranes and metal–boron bonding. Chem. Soc. Rev. 1974, 3, 231–271. [Google Scholar] [CrossRef]
- Grimes, R.N. Structure and stereochemistry in metalloboron cage compounds. Acc. Chem. Res. 1978, 11, 420–427. [Google Scholar] [CrossRef]
- Grimes, R.N. Metallacarboranes and metal-boron clusters in organometallic synthesis. Pure Appl. Chem. 1982, 54, 43–58. [Google Scholar] [CrossRef]
- Barton, L.; Srivastava, D.K. Comprehensive Organometallic Chemistry II; Abel, E., Stone, F.G.A., Wilkinson, G.E., Eds.; Pergamon: New York, NY, USA, 1995; Volume 1, pp. 275–373. [Google Scholar]
- Grimes, R.N. Metal Interactions with Boron Clusters; Plenum: New York, NY, USA, 1982; pp. 269–319. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Wang, X. Low-Valent Ditantalum Complex Ta2(μ-BH3)(μ-dmpm)3(η2-BH4)2: First Dinuclear Compound Containing a Bridging BH3 Group with Direct Ta–B Bonds. J. Am. Chem. Soc. 1998, 120, 9594–9599. [Google Scholar] [CrossRef]
- Kennedy, J.D. The Polyhedral Metallaboranes. Prog. Inorg. Chem. 1984, 32, 519–670. [Google Scholar]
- Kennedy, J.D. The polyhedral metallaboranes, Part II, Metallaborane clusters with eight vertices and more. Prog. Inorg. Chem. 1986, 34, 211–434. [Google Scholar]
- Kennedy, J.D. Disobedient Skeletons; Casanova, J., Ed.; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Pipal, J.R.; Grimes, R.N. Crystal structure of a tetracobalt tetraboron cluster, (η5-C5H5)4Co4B4H4. Structural patterns in eight-vertex polyhedra. Inorg. Chem. 1979, 18, 257–263. [Google Scholar] [CrossRef]
- Housecroft, C.E.; Fehlner, T.P. Metalloboranes: Their Relationships to Metal–Hydrocarbon Complexes and Clusters. Adv. Organomet. Chem. 1982, 21, 57–112. [Google Scholar] [CrossRef]
- Grimes, R.N. Boron Clusters Come of Age. J. Chem. Educ. 2004, 81, 657–672. [Google Scholar] [CrossRef]
- Grimes, R.N. Comprehensive Organometallic Chemistry II; Abel, E., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon: New York, NY, USA, 1995; Volume 1, Chapter 9. [Google Scholar]
- Gaines, D.F. Recent Advances in the Chemistry of Pentaborane (9); Parry, R.W., Kodama, G., Eds.; Pergamon: Oxford, UK, 1980. [Google Scholar]
- Shore, S.G.; Jan, D.-Y.; Hsu, L.-Y.; Hsu, W.-L. Insertion of boron into an osmium–carbonyl bond. Preparation and structure of the carbonyl borylidyne (μ-H)3(CO)9Os3BCO. J. Am. Chem. Soc. 1983, 105, 5923–5924. [Google Scholar] [CrossRef]
- Jan, D.-Y.; Workman, D.P.; Hsu, L.-Y.; Krause, J.A.; Shore, S.G. Clusters derived from the hydroboration of decacarbonyldi-.mu.-hydridotriosmium and their derivatives. Inorg. Chem. 1992, 31, 5123–5131. [Google Scholar] [CrossRef]
- Leyden, R.N.; Sullivan, B.P.; Baker, R.T.; Hawthrone, M.F. Synthesis of closo- and nido-metalloboranes from metallocenes. J. Am. Chem. Soc. 1978, 100, 3758–3765. [Google Scholar] [CrossRef]
- Lupan, A.; King, R.B. Boron in Organometallic Chemistry. In Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine; Hosmane, N.S., Eagling, R., Eds.; World Scientific: London, UK, 2018; Volume 1, Chapter 1; pp. 1–20. [Google Scholar]
- Hosmane, N.S. Boron Science: New Technologies and Applications; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2011. [Google Scholar]
- Weller, A.S. Comprehensive Organometallic Chemistry III; Mingos, D.M.P., Crabtree, R.H., Eds.; Pergamon: New York, NY, USA, 2007; Volume 3, Chapter 3.04; pp. 133–174. [Google Scholar]
- Hosmane, N.S.; Maguire, J.A. Comprehensive Organometallic Chemistry III; Mingos, D.M.P., Crabtree, R.H., Eds.; Pergamon: New York, NY, USA, 2007; Volume 3, Chapter 3.05; pp. 175–264. [Google Scholar]
- Bose, S.K.; Geetharani, K.; Varghese, B.; Mobin, S.M.; Ghosh, S. Metallaboranes of the Early Transition Metals: Direct Synthesis and Characterization of [{(η5-C5Me5)Ta}2BnHm] (n = 4, m = 10; n = 5, m = 11), [{(η5-C5Me5)Ta}2B5H10(C6H4CH3)], and [{(η5-C5Me5)TaCl}2B5H11]. Chem. Eur. J. 2008, 14, 9058–9064. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Reddy, K.H.K.; Dhayal, R.S.; Mobin, S.M.; Ramkumar, V.; Jemmis, E.D.; Ghosh, S. Chlorinated Hypoelectronic Dimetallaborane Clusters: Synthesis, Characterization, and Electronic Structures of (η5-C5Me5W)2B5HnClm (n = 7, m = 2 and n = 8, m = 1). Inorg. Chem. 2009, 48, 6509–6516. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Dhayal, R.S.; Varghese, B.; Ghosh, S. Unusual Open Eight-Vertex Oxamolybdaboranes: Structural Characterizations of (η5-C5Me5Mo)2B5(μ3-OEt)H6R (R = H and n-BuO). Organometallics 2009, 28, 1586–1589. [Google Scholar] [CrossRef]
- Geetharani, K.; Bose, S.K.; Pramanik, G.; Saha, T.K.; Ramkumar, V.; Ghosh, S. An Efficient Route to Group 6 and 8 Metallaborane Compounds: Synthesis of arachno-[Cp*Fe(CO)B3H8] and closo-[(Cp*M)2B5H9] (M = Mo, W). Eur. J. Inorg. Chem. 2009, 1483–1487. [Google Scholar] [CrossRef]
- Dhayal, R.S.; Chakrahari, K.K.V.; Ramkumar, V.; Ghosh, S. B-Alkylation and Arylation of [(η5-C5Me5Mo)2B5H9]: Synthesis and Characterization of Isomeric [(η5-C5Me5Mo)2B5H9−nRn] (When R = n-Bu, n = 2, 1; R = Ph, n = 2, 1). J. Clust. Sci. 2009, 20, 565–572. [Google Scholar] [CrossRef]
- Sahoo, S.; Mobin, S.M.; Ghosh, S. Direct insertion of sulfur, selenium and tellurium atoms into metallaborane cages using chalcogen powders. J. Organomet. Chem. 2010, 695, 945–949. [Google Scholar] [CrossRef]
- Bose, S.K.; Geetharani, K.; Ghosh, S. C–H activation of arenes and heteroarenes by early transition metallaborane, [(Cp*Ta)2B5H11] (Cp* = η5-C5Me5). Chem. Commun. 2011, 47, 11996–11998. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.K.; Bose, S.K.; Geetharani, K.; Chakrahari, K.K.V.; Mobin, S.M.; Ghosh, S. Synthesis and Structural Characterization of New Divanada- and Diniobaboranes Containing Chalcogen Atoms. Chem. Eur. J. 2012, 18, 9983–9991. [Google Scholar] [CrossRef] [PubMed]
- Chakrahari, K.K.; Thakur, A.; Mondal, B.; Ramkumar, V.; Ghosh, S. Hypoelectronic Dimetallaheteroboranes of Group 6 Transition Metals Containing Heavier Chalcogen Elements. Inorg. Chem. 2013, 52, 7923–7932. [Google Scholar] [CrossRef] [PubMed]
- Chakrahari, K.K.; Thakur, A.; Anju, V.P.; Ghosh, S. B–H bond iodination of polyhedral dimolybdaborane and dimolybdathiaborane clusters. J. Organomet. Chem. 2014, 751, 321–325. [Google Scholar] [CrossRef]
- Bag, R.; Mondal, B.; Bakthavachalam, K.; Roisnel, T.; Ghosh, S. Heterometallic boride clusters: Synthesis and characterization of butterfly and square pyramidal boride clusters. Pure Appl. Chem. 2018, 90, 665–675. [Google Scholar] [CrossRef]
- Hashimoto, H.; Shang, M.; Fehlner, T.P. Clusters as Ligands. Coordination of an Electronically Unsaturated Chromaborane to an Iron Tricarbonyl Fragment. J. Am. Chem. Soc. 1996, 118, 8164–8165. [Google Scholar] [CrossRef]
- Aldridge, S.; Hashimoto, H.; Kawamura, K.; Shang, M.; Fehlner, T.P. Cluster Expansion Reactions of Group 6 Metallaboranes. Syntheses, Crystal Structures, and Spectroscopic Characterizations of (Cp*Cr)2B5H9, (Cp*Cr)2B4H8Fe(CO)3, (Cp*Cr)2B4H7Co(CO)3, and (Cp*Mo)2B5H9Fe(CO)3. Inorg. Chem. 1998, 37, 928–940. [Google Scholar] [CrossRef]
- Aldridge, S.; Fehlner, T.P.; Shang, M. Directed Synthesis of Chromium and Molybdenum Metallaborane Clusters. Preparation and Characterization of (Cp*Cr)2B5H9, (Cp*Mo)2B5H9, and (Cp*MoCl)2B4H10. J. Am. Chem. Soc. 1997, 119, 2339–2340. [Google Scholar] [CrossRef]
- Aldridge, S.; Shang, M.; Fehlner, T.P. Synthesis of Novel Molybdaboranes from (η5-C5R5)MoCln Precursors (R = H, Me; n = 1, 2, 4). J. Am. Chem. Soc. 1998, 120, 2586–2598. [Google Scholar] [CrossRef]
- Kim, D.Y.; Girolami, G.S. Synthesis and Characterization of the Octahydrotriborate Complexes Cp*V(B3H8)2 and Cp*Cr(B3H8)2 and the Unusual Cobaltaborane Cluster Cp*2Co2(B6H14). J. Am. Chem. Soc. 2006, 128, 10969–10977. [Google Scholar] [CrossRef] [PubMed]
- Guennic, B.L.; Jiao, H.; Kahlal, S.; Saillard, J.-Y.; Halet, J.-F.; Ghosh, S.; Shang, M.; Beatty, A.M.; Rheingold, A.L.; Fehlner, T.P. Synthesis and Characterization of Hypoelectronic Rhenaboranes. Analysis of the Geometric and Electronic Structures of Species Following Neither Borane nor Metal Cluster Electron-Counting Paradigms. J. Am. Chem. Soc. 2004, 126, 3203–3217. [Google Scholar] [CrossRef] [PubMed]
- Weller, A.S.; Shang, M.; Fehlner, T.P. Synthesis of Mono- and Ditungstaboranes from Reaction of Cp*WCl4 and [Cp*WCl2]2 with BH3·thf or LiBH4 (Cp* = η5-C5Me5). Control of Reaction Pathway by Choice of Monoboron Reagent and Oxidation State of Metal Center. Organometallics 1999, 18, 53–64. [Google Scholar] [CrossRef]
- Bullick, H.J.; Grebenik, P.D.; Green, M.L.H.; Hughes, A.K.; Leach, J.B.; McGowan, P.C. Synthesis of η-cyclopentadienyl-polyborane derivatives of molybdenum and tungsten. J. Chem. Soc. Dalton Trans. 1995, 67–75. [Google Scholar] [CrossRef]
- Aldridge, S.; Shang, M.; Fehlner, T.P. Origins of Unsaturation in Group 6 Metallaboranes. Synthesis, Crystal Structure, and Molecular Orbital Calculations for (Cp*MoCl)2B3H7 (Cp* = Pentamethylcyclopenta -dienyl). J. Am. Chem. Soc. 1997, 119, 11120–11121. [Google Scholar] [CrossRef]
- Aldridge, S.; Hashimoto, H.; Shang, M.; Fehlner, T.P. Cp*TaCl2B4H8: Synthesis, crystal structure and spectroscopic characterization of an air-stable, electronically unsaturated, chiral tantalaborane. J. Chem. Soc. Chem. Commun. 1998, 207–208. [Google Scholar] [CrossRef]
- Bose, S.K.; Geetharani, K.; Varghese, B.; Ghosh, S. Condensed Tantalaborane Clusters: Synthesis and Structures of [(Cp*Ta)2B5H7{Fe(CO)3}2] and [(Cp*Ta)2B5H9{Fe(CO)3}4]. Inorg. Chem. 2011, 50, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Geetharani, K.; Krishnamoorthy, B.S.; Kahlal, S.; Mobin, S.M.; Halet, J.-F.; Ghosh, S. Synthesis and Characterization of Hypoelectronic Tantalaboranes: Comparison of the Geometric and Electronic Structures of [(Cp*TaX)2B5H11] (X = Cl, Br, and I). Inorg. Chem. 2012, 51, 10176–10184. [Google Scholar] [CrossRef] [PubMed]
- Yuvaraj, K.; Roy, D.K.; Geetharani, K.; Mondal, B.; Anju, V.P.; Shankhari, P.; Ramkumar, V.; Ghosh, S. Chemistry of Homo- and Heterometallic Bridged-Borylene Complexes. Organometallics 2013, 32, 2705–2712. [Google Scholar] [CrossRef]
- Anju, R.S.; Saha, K.; Mondal, B.; Dorcet, V.; Roisnel, T.; Halet, J.-F.; Ghosh, S. Chemistry of Diruthenium Analogue of Pentaborane(9) With Heterocumulenes: Toward Novel Trimetallic Cubane-Type Clusters. Inorg. Chem. 2014, 53, 10527–10535. [Google Scholar] [CrossRef] [PubMed]
- Geetharani, K.; Bose, S.K.; Sahoo, S.; Varghese, B.; Mobin, S.M.; Ghosh, S. Cluster Expansion Reactions of Group 6 and 8 Metallaboranes Using Transition Metal Carbonyl Compounds of Groups 7–9. Inorg. Chem. 2011, 50, 5824–5832. [Google Scholar] [CrossRef] [PubMed]
- Mondal, B.; Bag, R.; Roisnel, T.; Ghosh, S. Use of Single Metal Fragments for Cluster Building: Synthesis, Structure and Bonding of Heterometalla-boranes. Inorg. Chem. 2019, 58, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Mondal, B.; Bag, R.; Ghorai, S.; Bakthavachalam, K.; Jemmis, E.D.; Ghosh, S. Synthesis, Structure, Bonding, and Reactivity of Metal Complexes Comprising Diborane(4) and Diborene(2): [{Cp*Mo(CO)2}2{μ-η2:η2-B2H4}] and [{Cp*M(CO)2}2B2H2M(CO)4], M = Mo, W. Angew. Chem. Int. Ed. 2018, 57, 8079–8083. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lei, X.; Cahill, C.L.; Fehlner, T.P. Symmetrical Scission of the Coordinated Tetraborane in [(Cp*ReH2)2B4H4] on CO Addition and Reassociation of the Coordinated Diboranes on H2 Loss. Angew. Chem. Int. Ed. 2000, 39, 2900–2902. [Google Scholar] [CrossRef]
- Wade, K. Structural and Bonding Patterns in Cluster Chemistry. Adv. Inorg. Chem. Radiochem. 1976, 18, 1–66. [Google Scholar] [CrossRef]
- Mingos, D.M.P. Polyhedral skeletal electron pair approach. Acc. Chem. Res. 1984, 17, 311–319. [Google Scholar] [CrossRef]
- Jemmis, E.D.; Balakrishnarajan, M.N.; Pancharatna, P.D. Electronic Requirements for Macropolyhedral Boranes. Chem. Rev. 2002, 102, 93–144. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.K.; Mondal, B.; De, A.; Panda, S.; Ghosh, S. Novel Neutral Zirconaborane [(Cp2Zr)2B5H11]: An arachno-B3H9 Analogue (Cp = η5-C5H5). Organometallics 2015, 34, 908–912. [Google Scholar] [CrossRef]
- De, A.; Zhang, Q.-F.; Mondal, B.; Cheung, L.F.; Kar, S.; Saha, K.; Varghese, B.; Wang, L.-S.; Ghosh, S. [(Cp2M)2B9H11] (M = Zr or Hf): Early transition metal ‘guarded’ heptaborane with strong covalent and electrostatic bonding. Chem. Sci. 2018, 9, 1976–1981. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Grimes, R.N. Polyhedral ferraboranes derived from the B5H8− ion. Analogs of ferrocene, hexaborane(10), and nido-B11H15. Inorg. Chem. 1979, 18, 3291–3294. [Google Scholar] [CrossRef]
- Ghosh, S.; Noll, B.C.; Fehlner, T.P. Borane Mimics of Classic Organometallic Compounds: [(Cp*Ru)B8H14(RuCp*)]0,+, Isoelectronic Analogues of Dinuclear Pentalene Complexes. Angew. Chem. Int. Ed. 2005, 44, 6568–6571. [Google Scholar] [CrossRef] [PubMed]
- Wilczynski, R.; Sneddon, L.G. Synthesis of two new cobaltaborane complexes: 1-(η5-C5H5)CoB5H9 and 2-(η5-C5H5)CoB9H13. Inorg. Chem. 1979, 18, 864–866. [Google Scholar] [CrossRef]
- Ghosh, S.; Noll, B.C.; Fehlner, T.P. Expansion of iridaborane clusters by addition of monoborane. Novel metallaboranes and mechanistic detail. Dalton Trans. 2008, 371–378. [Google Scholar] [CrossRef]
- Dhayal, R.S.; Sahoo, S.; Ramkumar, V.; Ghosh, S. Substitution at boron in molybdaborane frameworks: Synthesis and characterization of isomeric (η-C5Me5Mo)2B5HnXm (when X = Cl: n = 5, 7, 8; m = 4, 2, 1 and X = Me: n = 6, 7; m = 3, 2). J. Organomet. Chem. 2009, 694, 237–243. [Google Scholar] [CrossRef]
- Green, M.L.H.; Hubert, J.D.; Mountford, P. Synthesis of the W≡W triply bonded dimers [W2(η5-C5H4R)2X4] (X = Cl, R = Me or Pri; X = Br, R = Pri) and X-ray crystal structures of [W(η5-C5H4Pri)Cl4] and [W2(η-C5H4Pri)2Cl4]. J. Chem. Soc. Dalton Trans. 1990, 3793–3800. [Google Scholar] [CrossRef]
- Kaushika, M.; Singh, A.; Kumar, M. The chemistry of group-VIb metal carbonyls. Eur. J. Chem. 2012, 3, 367–394. [Google Scholar] [CrossRef] [Green Version]
- Ryschkewitsch, G.E.; Nainan, K.C. Octahydrotriborate(1−) [B3H8] salt. Inorg. Synth. 1974, 15, 113–114. [Google Scholar] [CrossRef]
- Bruker. APEX2, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Sheldrick, G.M. SHELXS-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metallaborane | Avg.d [M–M] (Å) | Avg.d [M–B] (Å) | Avg.d [B–B] (Å) | 11B NMR (ppm) | Ref. |
|---|---|---|---|---|---|
| [(Cp*Cr)2B4H8Fe(CO)3] | 2.71 | 2.15 | 1.72 | - | [34] |
| [(Cp*Cr)2B4H7Co(CO)3] | 2.69 | 2.13 | 1.69 | - | [35] |
| [(Cp*W)2B5H5(μ-H)4] | 2.82 | 2.26 | 1.71 | 49.2, 46.9, 26.8 | [40] |
| [(Cp*Mo)2(μ-H)2(μ3-H)2B4H4W(CO)4] | 2.91 | 2.24 | 1.72 | 27.9, 41.9, 81.2 | [49] |
| [(Cp*Mo)2(μ-H)2B4H4W(CO)5] | 2.91 | 2.24 | 1.73 | 103.0, 77.9, 77.0 | [49] |
| [(Cp*W)2(μ-H)2(μ3-H)2B4H4Cr(CO)4], 1 | 2.84 | 2.38 | 1.73 | 70.6, 62.2, 36.5, 25.0 | This work |
| [(Cp*W)2(μ-H)2(μ3-H)2B4H4Mo(CO)4], 2 | 2.83 | 2.27 | 1.71 | 70.1, 60.1, 39.1, 25.6 | This work |
| [(Cp*W)2(μ-H)2(μ3-H)2B4H4W(CO)4], 3 | 2.89 | 2.24 | 1.71 | 70.2, 60.1, 35.5, 26.1 | This work |
| [(Cp*W)2(μ-H)2(μ3-H)2B4H4Fe2(CO)3], 4 | 2.82 | 2.28 | 1.74 | 79.0, 76.2, 41.0, 35.9 | This work |
| [(Cp*Mo)2(μ-H)2(μ3-H)2B4H4Cr(CO)4], 5 | 2.82 | 2.23 | 1.72 | 83.5, 81.3, 41.3, 27.1 | This work |
| Metallaborane | Geometry | Sep a | Avg.d [M–M] (Å) | Avg.d [M–B] (Å) | Avg.d [B–B] (Å) | 11B NMR (ppm) | Ref. |
|---|---|---|---|---|---|---|---|
| [(Cp2M)2B5H11] M = Zr, Hf |  | 10 | b | 2.50(Zr) 2.50(Hf) | 1.78(Zr) 1.78(Hf) | 8.1, 2.5, −4.1 (Zr) −3.8, 2.0, 4.6 (Hf) | [55,56] |
| [M2B5H11] M = CpV, Cp*Ta |  | 6 | 2.76 (V) 2.92 (Ta) | 2.23 (V) 2.32 (Ta) | 1.73 (V) 1.80 (Ta) | −1.8, 3.4, 21.9 (V) 3.7, 23.9, 44.7 (Ta) | [23,30] |
| [Cp*TaB5H11Cl2] |  | 8 | 3.22 | 2.38 | 1.76 | 77.7, 18.8, 15, −10 | [23] |
| [(Cp*M)2B5H9] M = Cr, Mo, W |  | 6 | 2.62 (Cr) 2.80 (Mo) 2.81 (W) | 2.15 (Cr) 2.24 (Mo) 2.23 (W) | 1.67 (Cr) 1.72 (Mo) 1.71 (W) | 25.0, 86.2, 91.5 (Cr) 62.9 (3 B), 25.8 (Mo) 26.8, 46.9, 49.2 (W) | [36,37,38,40] |
| [(Cp*Mo)2B4EH5X] E = S, Se; X = H E = Te; X = Cl |  | 6 | 2.78 (S) 2.77 (Se) 2.84 (Te) | 2.21 (S) 2.24 (Se) 2.17 (Te) | 1.67 (S) 1.83 (Se) 1.69 (Te) | 101.1, 76.7, 40.3, 10.1 (S) 100.7, 76.7, 41.8, 16.8 (Se) 95.5, 73.2, 40.7, 26.7 (Te) | [31] |
| [(Cp*Cr)2B4H8M] M = Fe(CO)3, Co.(CO)3 |  | 6 | 2.70 (Fe) 2.69 (Co.) | 2.15 (Fe) 2.15 (Co.) | 1.71 (Fe) 1.69 (Co.) | 27.3, 80.0, 117.5 (Fe) 36.6, 57.5, 113.9, 121.7 (Co.) | [35] |
| [(Cp*Mo)2B4H4M] M = H2Cr(CO)4 (5), H2W(CO)4, W(CO)5 |  | 6 | 2.82 (Cr) 2.82 (W) 2.83 (W) | 2.27 (Cr) 2.23 (W) 2.23 (W) | 1.72 (Cr) 1.71(W) 1.72 (W) | 83.5, 81.3, 41.3, 27.1 (Cr) 27.9, 41.9, 81.2 (W) 77.0, 77.9, 103.0 (W) | This Work, [49] |
| [(Cp*W)2B4H4M] M = Cr(CO)4 (1), Mo(CO)4 (2), W(CO)4 (3) |  | 6 | 2.84 (Cr) 2.83 (Mo) 2.89 (W) | 2.38 (Cr) 2.27 (Mo) 2.24 (W) | 1.73 (Cr) 1.71 (Mo) 1.71 (W) | 70.6, 62.2, 36.5, 25.0 (Cr) 70.1, 60.1, 39.1, 25.6 (Mo) 70.2, 60.1, 35.5, 26.1 (W) | This work |
| [(Cp*W)2B4H8M] M = Fe(CO)3 (4) |  | 6 | 2.82 | 2.28 | 1.74 | 79.0, 76.2, 41.0, 35.9 | This work |
| [(Cp*Re)2B5Cl5H2] |  | 6 | 2.76 | 2.20 | 1.74 | 28.1, 48.3, 88.3 | [39] |
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Empirical formula | C24H37B4O4CrW2 | C24H30B4O4MoW2 | C24H30B4O4W3 | C23H38B4O3FeW2 | C24H38B4O4CrMo2 |
| Formula weight | 852.47 | 889.36 | 977.27 | 829.32 | 677.66 |
| Crystal system | Orthorhombic | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | Pca21 | P−1 | P−1 | P21/n | P21/c |
| a (Å) | 23.323(5) | 16.3096(16) | 16.8420(15) | 11.6885(13) | 19.454(2) |
| b (Å) | 14.555(3) | 18.8186(18) | 19.051(2) | 16.2795(16) | 13.2430(11) |
| c (Å) | 17.009(7) | 19.9272(18) | 19.390(2) | 15.0773(17) | 21.905(2) |
| α (°) | 90 | 85.532(3) | 85.443(4) | 90 | 90 |
| β (°) | 90.000(10) | 71.947(3) | 79.671(3) | 110.538(4) | 94.494(4) |
| γ (°) | 90 | 89.111(3) | 70.858(3) | 90 | 90 |
| V(Å3) | 5774(3) | 5797.1(10) | 5780.6(10) | 2686.6(5) | 5626.2(9) |
| Z | 8 | 8 | 8 | 4 | 8 |
| Dcalc (g/cm3) | 1.961 | 2.038 | 2.246 | 2.050 | 1.600 |
| F (000) | 3240 | 3328 | 3584 | 1576 | 2736 |
| µ (mm−1) | 8.346 | 8.370 | 11.937 | 9.097 | 1.285 |
| θ Range (°) | 3.2–22.1 | 2.27–27.51 | 2.26–27.50 | 2.50–27.52 | 2.57–27.46 |
| no. of reflns collected | 8977 | 26525 | 26460 | 6147 | 12768 |
| no. of unique reflns [I > 2σ(I)] | 3645 | 22650 | 22550 | 5606 | 10014 |
| goodness-of-fit on F2 | 0.997 | 1.065 | 1.104 | 1.063 | 1.018 |
| final R indices [I > 2σ(I)] | R1 = 0.0879, wR2 = 0.1580 | R1 = 0.0349, wR2 = 0.0819 | R1 = 0.0965, wR2 = 0.1988 | R1 = 0.0379, wR2 = 0.1175 | R1= 0.0810, wR2 = 0.1827 |
| R indices (all data) | R1 = 0.2386, wR2 = 0.2215 | R1 = 0.0458, wR2 = 0.0880 | R1 = 0.1133, wR2 = 0.2103 | R1 = 0.0421, wR2 = 0.1236 | R1 = 0.1054, wR2 = 0.2014 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bag, R.; Saha, S.; Borthakur, R.; Mondal, B.; Roisnel, T.; Dorcet, V.; Halet, J.-F.; Ghosh, S. Synthesis, Structures and Chemistry of the Metallaboranes of Group 4–9 with M2B5 Core Having a Cross Cluster M–M Bond. Inorganics 2019, 7, 27. https://doi.org/10.3390/inorganics7030027
Bag R, Saha S, Borthakur R, Mondal B, Roisnel T, Dorcet V, Halet J-F, Ghosh S. Synthesis, Structures and Chemistry of the Metallaboranes of Group 4–9 with M2B5 Core Having a Cross Cluster M–M Bond. Inorganics. 2019; 7(3):27. https://doi.org/10.3390/inorganics7030027
Chicago/Turabian StyleBag, Ranjit, Suvam Saha, Rosmita Borthakur, Bijan Mondal, Thierry Roisnel, Vincent Dorcet, Jean-François Halet, and Sundargopal Ghosh. 2019. "Synthesis, Structures and Chemistry of the Metallaboranes of Group 4–9 with M2B5 Core Having a Cross Cluster M–M Bond" Inorganics 7, no. 3: 27. https://doi.org/10.3390/inorganics7030027
APA StyleBag, R., Saha, S., Borthakur, R., Mondal, B., Roisnel, T., Dorcet, V., Halet, J. -F., & Ghosh, S. (2019). Synthesis, Structures and Chemistry of the Metallaboranes of Group 4–9 with M2B5 Core Having a Cross Cluster M–M Bond. Inorganics, 7(3), 27. https://doi.org/10.3390/inorganics7030027