Comparative Study of Complexes of Rare Earths and Actinides with 2,6-Bis(1,2,4-triazin-3-yl)pyridine


Abstract
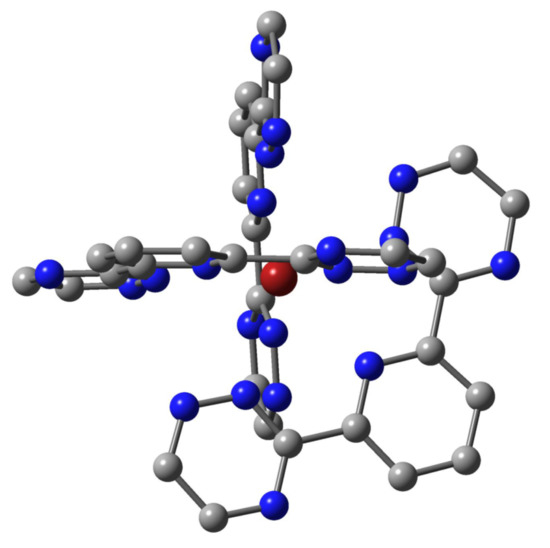
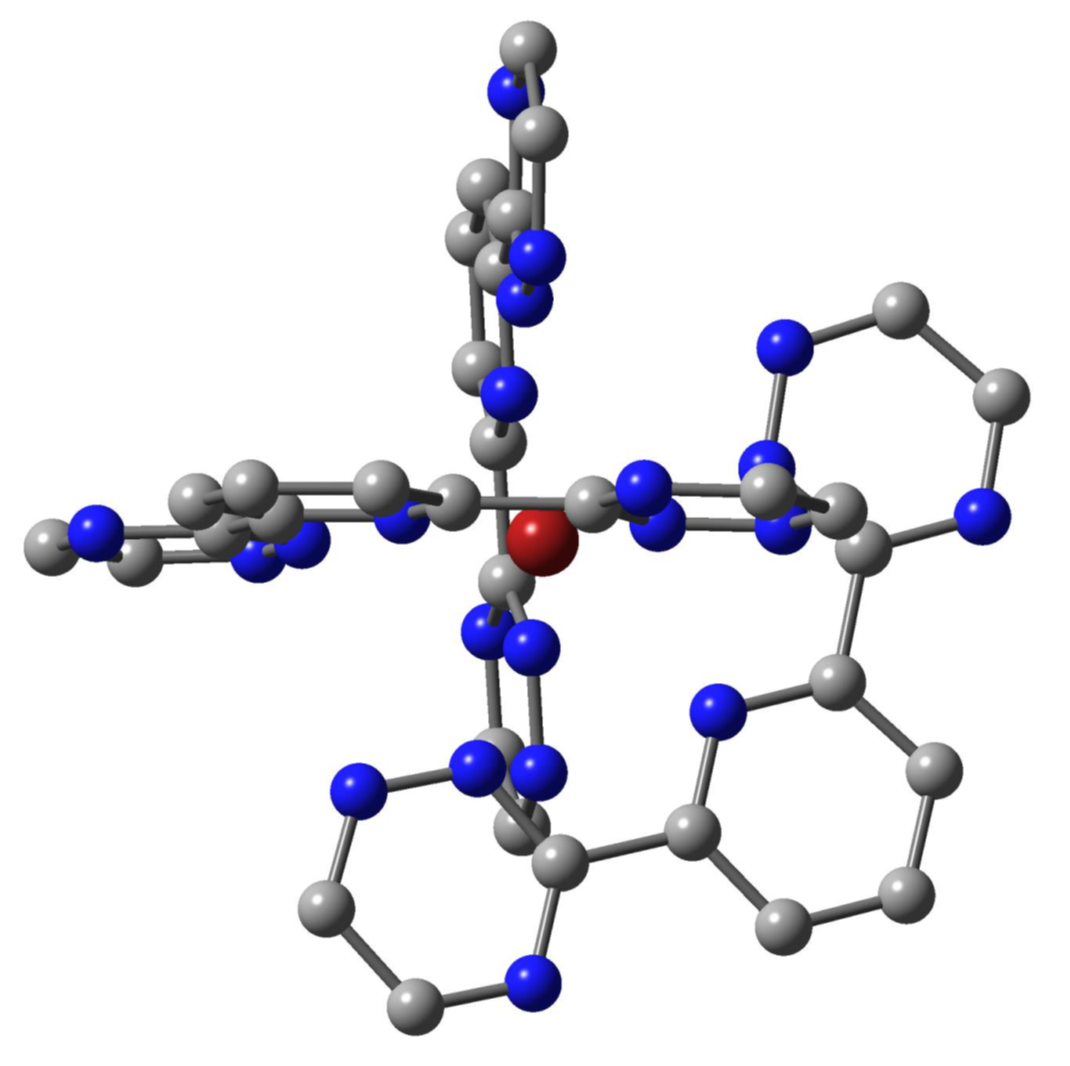
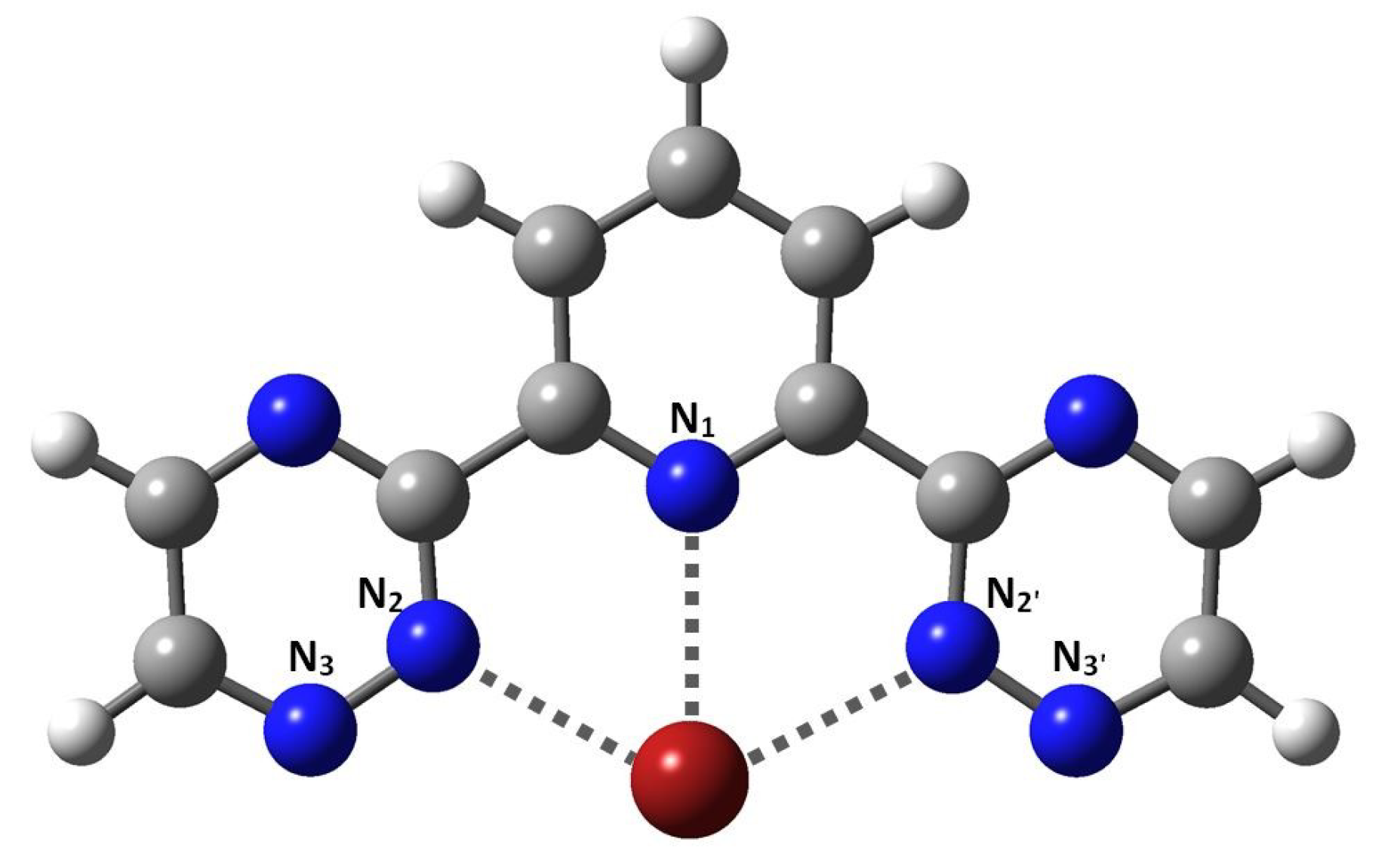
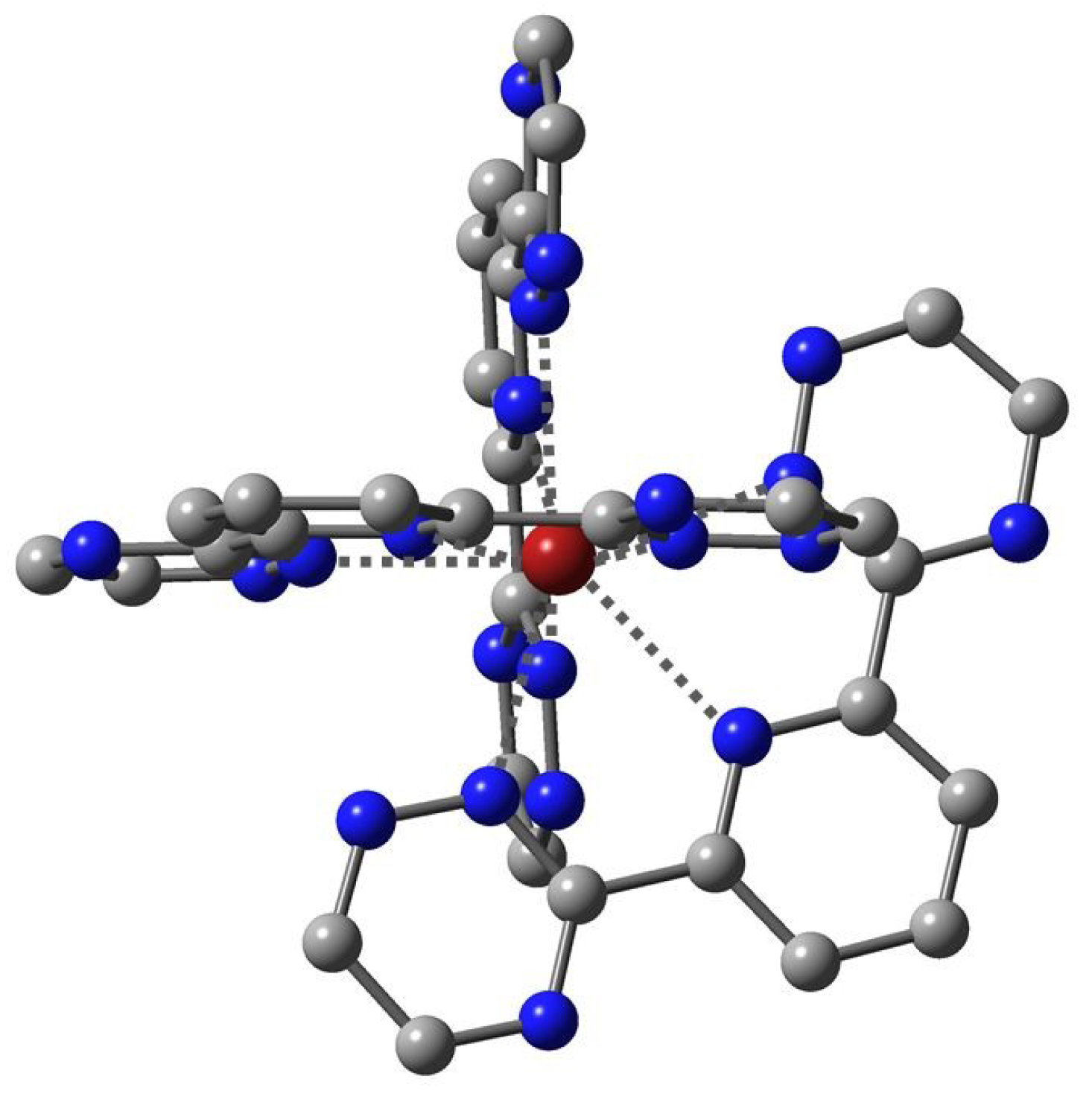
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Preparation Procedure of the Complexes [M(BTP)3](CF3SO3)3
3.2. Crystallography
3.3. IR Spectroscopy
3.4. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kolarik, Z.; Müllich, U.; Gassner, F. Selective Extraction Of Am(III) Over Eu(III) by 2,6-Ditriazolyl- and 2,6-Ditriazinylpyridines. Solvent Extr. Ion Exch. 1999, 17, 23–32. [Google Scholar] [CrossRef]
- Kolarik, Z.; Müllich, U.; Gassner, F. Extraction of Am(III) and Eu(III) Nitrates by 2-6-Di-(5,6-Dipropyl-1,2,4-Triazin-3-yl)Pyridines. Solvent Extr. Ion Exch. 1999, 17, 1155–1170. [Google Scholar] [CrossRef]
- Berthet, J.C.; Miquel, Y.; Iveson, P.B.; Nierlich, M.; Thuery, P.; Madic, C.; Ephritikhine, M. The affinity and selectivity of terdentate nitrogen ligands towards trivalent lanthanide and uranium ions viewed from the crysatl structures of the 1:3 complexes. J. Chem. Soc. Dalton Trans. 2002, 16, 3265–3272. [Google Scholar] [CrossRef]
- Choppin, G.; Liljenzin, J.-O.; Rydberg, J. Radiochemistry and Nuclear Chemistry; Butterworth-Heinemann: Woburn, MA, USA, 2002. [Google Scholar]
- Geist, A.; Hill, C.; Modolo, G.; Foreman, M.R.S.J.; Weigl, M.; Gompper, K.; Hudson, M.J. 6,6′-Bis(5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-benzo[1,2,4]triazin-3-yl) [2,2′]bipyridine, an Effective Extracting Agent for the Separation of Americium(III) and Curium(III) from the Lanthanides. Solv. Extr. Ion Exch. 2006, 24, 463–483. [Google Scholar] [CrossRef]
- Lan, J.-H.; Shi, W.-Q.; Yuan, L.-Y.; Li, J.; Zhao, Y.-L.; Chai, Z.-F. Recent advances in computational modeling and simulations on the An(III)/Ln(III) separation process. Coord. Chem. Rev. 2012, 256, 1406–1417. [Google Scholar] [CrossRef]
- Neidig, M.L.; Clark, D.L.; Martin, R.L. Covalency in f-element complexes. Coord. Chem. Rev. 2013, 257, 394–406. [Google Scholar] [CrossRef]
- Benay, G.; Schurhammer, R.; Desaphy, J.; Wipff, G. Substituent effects on BTP’s basicity and complexation properties with LnIII lanthanide ions. New J. Chem. 2011, 35, 184–189. [Google Scholar] [CrossRef]
- Trumm, S.; Wipff, G.; Geist, A.; Panak, P.J.; Fanghänel, T. Optimising BTP ligands by tuning their basicity. Radiochim. Acta Int. J. Chem. Asp. Nucl. Sci. Technol. 2011, 99, 13–16. [Google Scholar] [CrossRef]
- Ionova, G.; Rabbe, C.; Guillaumont, R.; Ionov, S.; Madic, C.; Krupac, J.-C.; Guillaneux, D. A donor–acceptor model of Ln(III) complexation with terdentate nitrogen planar ligands. New J. Chem. 2002, 26, 234–242. [Google Scholar] [CrossRef]
- Panak, P.J.; Geist, A. Complexation and Extraction of Trivalent Actinides and Lanthanides by Triazinylpyridine N-Donor Ligands. Chem. Rev. 2013, 113, 1199–1236. [Google Scholar] [CrossRef] [PubMed]
- Iveson, P.B.; Rivière, C.; Guillaneux, D.; Niederlich, M.; Thuéry, P.; Epheritikine, M.; Madic, C. Selective complexation of uranium(III) over cerium(III) by 2,6-bis(5,6-dialkyl-1,2,4-triazin-3-yl)pyridines: 1H NMR and X-ray crystallography studies. Chem. Commun. 2001, 16, 1512–1513. [Google Scholar] [CrossRef]
- Guillaumont, D. Quantum Chemistry Study of Actinide(III) and Lanthanide(III) Complexes with Tridentate Nitrogen Ligands. J. Phys. Chem. A 2004, 108, 6893–6900. [Google Scholar] [CrossRef]
- Denecke, M.A.; Rossberg, A.; Panak, P.J.; Weigl, M.; Schimmelpfennig, B.; Geist, A. Characterization and comparison of Cm(III) and Eu(III) complexed with 2,6-di(5,6-dipropyl-1,2,4-triazin-3-yl)pyridine using EXAFS, TRFLS, and quantum-chemical methods. Inorg. Chem. 2005, 44, 8418–8425. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Adamo, C.; Maldivi, P. Toward a Clear-Cut Vision on the Origin of 2,6-Di(1,2,4-triazin-3-yl)pyridine Selectivity for Trivalent Actinides: Insights from Theory. Inorg. Chem. 2006, 45, 8517–8522. [Google Scholar] [CrossRef] [PubMed]
- Maldivi, P.; Petit, L.; Adamo, C.; Vetere, V. Theoretical description of metal-ligand bonding within f-element complexes: A successful and necessary interplay between theory and experiment. C. R. Chimie 2007, 10, 888–896. [Google Scholar] [CrossRef]
- Steppert, M.; Walther, C.; Geist, A.; Fanghänel, T. Direct nano ESI time-of-flight mass spectrometric investigations on lanthanide BTP complexes in the extraction-relevant diluent 1-octanol. New J. Chem. 2009, 33, 2437–2442. [Google Scholar] [CrossRef]
- Kolarik, Z. Complexation and Separation of Lanthanides(III) and Actinides(III) by Heterocyclic N-Donors in Solutions. Chem. Rev. 2008, 108, 4208–4252. [Google Scholar] [CrossRef] [PubMed]
- Banik, N.L.; Schimmelpfennig, B.; Marquard, C.M.; Brendebach, B.; Geist, A.; Denecke, M.A. Characterization of redox sensitive plutonium(III) complexed with alkylated 2,6-ditriazinylpyridine (BTP) in organic solution. Dalton Trans. 2010, 39, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Hill, C.; Suzenet, F.; Guillaumet, G. Influence of an Alkoxy Group on Bis-Triazinyl-Pyridines for Selective Extraction of Americium(III). Solv. Extract. Ion Exch. 2013, 31, 253–268. [Google Scholar] [CrossRef]
- Lan, J.-H.; Shi, W.-Q.; Yuan, L.-Y.; Zhao, Y.-L.; Li, J.; Chai, Z.-F. Trivalent Actinide and Lanthanide Separations by Tetradentate Nitrogen Ligands: A Quantum Chemistry Study. Inorg. Chem. 2011, 50, 9230–9237. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.-H.; Shi, W.-Q.; Yuan, L.-Y.; Feng, Y.-X.; Zhao, Y.-L.; Chai, Z.-F. Thermodynamic Study on the Complexation of Am(III) and Eu(III) with Tetradentate Nitrogen Ligands: A Probe of Complex Species and Reactions in Aqueous Solution. J. Phys. Chem. A 2012, 116, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Narbutt, J.; Oziminski, W.P. Selectivity of bis-triazinyl bipyridine ligands for americium(III) in Am/Eu separation by solvent extraction. Part 1. Quantum mechanical study on the structures of BTBP complexes and on the energy of the separation. Dalton Trans. 2012, 41, 14416–14424. [Google Scholar] [CrossRef] [PubMed]
- Narbutt, J.; Wodyński, A.; Pecul, M. The selectivity of diglycolamide (TODGA) and bis-triazine-bipyridine (BTBP) ligands in actinide/lanthanide complexation and solvent extraction separation-a theoretical approach. Dalton Trans. 2015, 44, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Rauk, A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta 1977, 46, 1–10. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Kovács, A.; Apostolidis, C.; Walter, O.; Lindqvist-Reis, P. “Lanthanide contraction” in [Ln(BTP)3](CF3SO3)3 complexes. Struct. Chem. 2015, 26, 1287–1295. [Google Scholar] [CrossRef]
- Fryer-Kanssen, I.; Austin, J.; Kerridge, A. Topological Study of Bonding in Aquo and Bis(triazinyl)pyridine Complexes of Trivalent Lanthanides and Actinides: Does Covalency Imply Stability? Inorg. Chem. 2016, 55, 10034–10042. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Drew, M.G.B.; Guillaneux, D.; Hudson, M.J.; Iveson, P.B.; Russell, M.L.; Madic, C. Lanthanide(III) complexes of a highly efficient actinide(III) extracting agent—2,6-bis(5,6-dipropyl-1,2,4-triazin-3-yl)pyridine. Inorg. Chem. Commun. 2001, 4, 12–15. [Google Scholar] [CrossRef]
- Fedosseev, A.M.; Grigoriev, M.S.; Charushnikova, I.A.; Budantseva, N.A.; Starikova, Z.A.; Moisy, P. Synthesis, crystal structure and some properties of new perrhenate and pertechnetate complexes of Nd3+ and Am3+ with 2,6-bis(tetramethylfurano)-1,2,4-triazin-3-yl)-pyridine, tris(2-pyridylmethyl)amine and N,N′-tetraethylmalonamide. Polyhedron 2008, 27, 2007–2014. [Google Scholar] [CrossRef]
- Guillet, G.L.; Hyatt, I.F.D.; Hillesheim, P.C.; Abboud, K.A.; Scott, M.J. 1,2,4-Triazine-picolinamide functionalized, nonadentate chelates for the segregation of lanthanides(III) and actinides(III) in biphasic systems. New J. Chem. 2013, 37, 119–131. [Google Scholar] [CrossRef]
- Frost, G.H.; Hart, F.A.; Heath, C.; Hursthouse, M.B. The crystal structure of tris-(2,2′,6′,2″-terpyridyl)europium(III) perchlorate. J. Chem. Soc. D 1969, 23, 1421–1422. [Google Scholar] [CrossRef]
- Piquet, C.; Williams, A.F.; Bernardinelli, G.; Bunzli, J.-C. Structural and photophysical properties of lanthanide complexes with planar aromatic tridentate nitrogen ligands as luminescent building blocks for triple-helical structures. Inorg. Chem. 1993, 32, 4139–4149. [Google Scholar] [CrossRef]
- Mallet, C.; Thummel, R.P.; Hery, C. Conformational properties of Eu(III) complexes of 3,3′; 5,3″-polymethylene bridged derivatives of 2,2′; 6,2″-terpyridine. Inorg. Chim. Acta 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Piguet, C.; Bunzli, J.-C.G.; Bernardinelli, G.; Bochet, C.G.; Froidevaux, P. Design of luminescent building blocks for supramolecular triple-helical lanthanide complexes. J. Chem. Soc. Dalton Trans. 1995, 1, 83–97. [Google Scholar] [CrossRef]
- Bardwell, D.A.; Jeffery, J.C.; Jones, P.L.; McCleverty, J.A.; Psillakis, E.; Reeves, Z.; Ward, M.D. Lanthanide complexes of the tetradentate N-donor ligand dihydrobis[3-(2-pyridyl)pyrazolyl]borate and the terdentate N-donor ligand 2,6-bis(1H-pyrazol-3-yl)pyridine: Syntheses, crystal structures and solution structures based on luminescence lifetime studies. J. Chem. Soc. Dalton Trans. 1997, 12, 2079–2086. [Google Scholar]
- Semenova, L.I.; Sobolev, A.N.; Skelton, B.W.; White, A.H. Structural Systematics of Rare Earth Complexes. XV Tris(2,2′:6′,2″-terpyridine)lanthanoid(III) Tris(perchlorate) Complexes. Aust. J. Chem. 1999, 52, 519–529. [Google Scholar] [CrossRef]
- Li, Y.; Huffman, J.C.; Flood, A.H. Can terdentate 2,6-bis(1,2,3-triazol-4-yl)pyridines form stable coordination compounds? Chem. Commun. 2007, 26, 2692–2694. [Google Scholar] [CrossRef] [PubMed]
- Meudtner, R.M.; Ostermeier, M.; Goddard, R.; Limberg, C.; Hecht, S. Multifunctional “Clickates” as Versatile Extended Heteroaromatic Building Blocks: Efficient Synthesis via Click Chemistry, Conformational Preferences, and Metal Coordination. Chem. Eur. J. 2007, 13, 9834–9840. [Google Scholar] [CrossRef] [PubMed]
- de Bettencourt-Dias, A.; Barber, P.S.; Viswanathan, S.; de Lill, D.T.; Rollett, A.; Ling, G.; Altun, S. Para-Derivatized Pybox Ligands As Sensitizers in Highly Luminescent Ln(III) Complexes. Inorg. Chem. 2010, 49, 8848–8861. [Google Scholar] [CrossRef] [PubMed]
- Yip, Y.-W.; Wen, H.; Wong, W.-T.; Tanner, P.A.; Wong, K.-L. Increased Antenna Effect of the Lanthanide Complexes by Control of a Number of Terdentate N-Donor Pyridine Ligands. Inorg. Chem. 2012, 51, 7013–7015. [Google Scholar] [CrossRef] [PubMed]
- Andreiadis, E.S.; Imbert, D.; Pecaut, J.; Demadrille, R.; Mazzanti, M. Self-assembly of highly luminescent lanthanide complexes promoted by pyridine-tetrazolate ligands. Dalton Trans. 2012, 41, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Shavaleev, N.M.; Eliseeva, S.V.; Scopelliti, R.; Bunzli, J.-C.G. Tridentate Benzimidazole-Pyridine-Tetrazolates as Sensitizers of Europium Luminescence. Inorg. Chem. 2014, 53, 5171–5178. [Google Scholar] [CrossRef] [PubMed]
- WebElements. Available online: http://www.webelements.com (accessed on 25 February 2019).
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Huheey, J.E.; Keiter, E.A.; Keiter, R.L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th ed.; HarperCollins: New York, NY, USA, 1993. [Google Scholar]
- Shannon, R.D.; Prewitt, C.T. Revised values of effective ionic radii. Acta Cryst. 1970, B26, 1046–1048. [Google Scholar] [CrossRef]
- Paul, P.; Ghosh, M.; Neogy, D.; Mallick, P.K. Electronic and vibrational spectra of some rare earth trifluoromethanesulfonates crystals. Spectrochim. Acta A 2011, 78, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Mann, J.B. Atomic Structure Calculations II. Hartree-Fock Wave Functions and Radial Expectation Values: Hydrogen to Lawrencium; LA-3691; Los Alamos Scientific Laboratory: Los Alamos, NM, USA, 1968. [Google Scholar]
- Kerridge, A. Oxidation state and covalency in f-element metallocenes (M = Ce, Th, Pu): A combined CASSCF and topological study. Dalton Trans. 2013, 42, 16428–16436. [Google Scholar] [CrossRef] [PubMed]
- Kerridge, A. f-Orbital covalency in the actinocenes (An = Th–Cm): Multiconfigurational studies and topological analysis. RSC Adv. 2014, 4, 12078–12086. [Google Scholar] [CrossRef]
- Di Pietro, P.; Kerridge, A. U–Oyl Stretching Vibrations as a Quantitative Measure of the Equatorial Bond Covalency in Uranyl Complexes: A Quantum-Chemical Investigation. Inorg. Chem. 2016, 55, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Kaltsoyannis, N. Covalency hinders AnO2(H2O)+→AnO(OH)2+ isomerisation (An = Pa–Pu). Dalton Trans. 2016, 45, 3158–3162. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, P.; Kerridge, A. Ligand size dependence of U–N and U–O bond character in a series of uranyl hexaphyrin complexes: Quantum chemical simulation and density based analysis. Phys. Chem. Chem. Phys. 2017, 19, 7546–7559. [Google Scholar] [CrossRef] [PubMed]
- Kerridge, A. Quantification of f-element covalency through analysis of the electron density: Insights from simulation. Chem. Commun. 2017, 53, 6685–6695. [Google Scholar] [CrossRef] [PubMed]
- Tanti, J.; Lincoln, M.; Kerridge, A. Decomposition of d- and f-shell contributions to uranium bonding from the quantum theory of atoms in molecules: Application to uranium and uranyl halides. Inorganics 2018, 6, 88. [Google Scholar] [CrossRef]
- Kaltsoyannis, N. Transuranic Computational Chemistry. Chem. Eur. J. 2018, 24, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, B.; Mishra, V. The coordination chemistry of lanthanide and actinide metal ions with hydroxypyridinone-based decorporation agents: Orbital and density based analyses. Dalton Trans. 2018, 47, 16603–16615. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Cheng, C.; Su, J.; Huai, P. Bonding nature of the actinide tetrafluorides AnF4 (An = Th–Cm). Mol. Phys. 2015, 113, 3450–3458. [Google Scholar] [CrossRef]
- Jensen, M.P.; Bond, A.H. Comparison of Covalency in the Complexes of Trivalent Actinide and Lanthanide Cations. J. Am. Chem. Soc. 2002, 124, 9870–9877. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.P.; King, F.L.; Goeringer, D.E.; Duckworth, D.C. Collision-induced dissociation of lanthanide oxide ions in quadrupole ion traps: Effects of bond strength and mass. Int. J. Mass Spectrom. 2002, 2016, 85–93. [Google Scholar] [CrossRef]
- Konings, R.J.M.; Beneš, O.; Kovács, A.; Manara, D.; Sedmidubský, D.; Gorokhov, L.; Iorish, V.S.; Yungman, V.; Shenyavskaya, E.; Osina, E. The thermodynamic properties of the f-elements and their compounds. Part II. The Lanthanide and Actinide Oxides. J. Phys. Chem. Ref. Data 2014, 43, 013101. [Google Scholar] [CrossRef]
- SMART, SAINT, SADABS; Siemens, Analytical X-ray Instruments Inc.: Karlsruhe, Germany, 1997.
- APEX2, SAINT-Plus, SADABS; Bruker AXS Inc.: Madison, WI, USA,, 2007.
- Sheldrick, G.M. SHELX-2013 programme package for structure solution and refinement. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar]
- Martin, J.M.L.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef] [Green Version]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Savin, A.; Preuss, H. Energy-adjusted pseudopotentials for the rare earth elements. Theor. Chim. Acta 1989, 75, 173–194. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds. Theor. Chim. Acta 1993, 85, 441–450. [Google Scholar] [CrossRef]
- Yang, J.; Dolg, M. Valence basis sets for lanthanide 4f-in-core pseudopotentials adapted for crystal orbital ab initio calculations. Theor. Chem. Acc. 2005, 113, 212–224. [Google Scholar] [CrossRef]
- Weigand, A.; Cao, X.; Yang, J.; Dolg, M. Quasirelativistic f-in-core pseudopotentials and core-polarization potentials for trivalent actinides and lanthanides: Molecular test for trifluorides. Theor. Chem. Acc. 2009, 126, 117–127. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-Adjusted Pseudopotentials for the Actinides. Parameter Sets and Test Calculations for Thorium and Thorium Monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M.; Stoll, H. Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials. J. Chem. Phys. 2003, 118, 487–496. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M. Segmented contraction scheme for small-core actinide pseudopotential basis sets. J. Mol. Struct. 2004, 673, 203–209. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.9; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2011. [Google Scholar]
- Keith, T.A. AIMAll (Version 17.11.14); TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
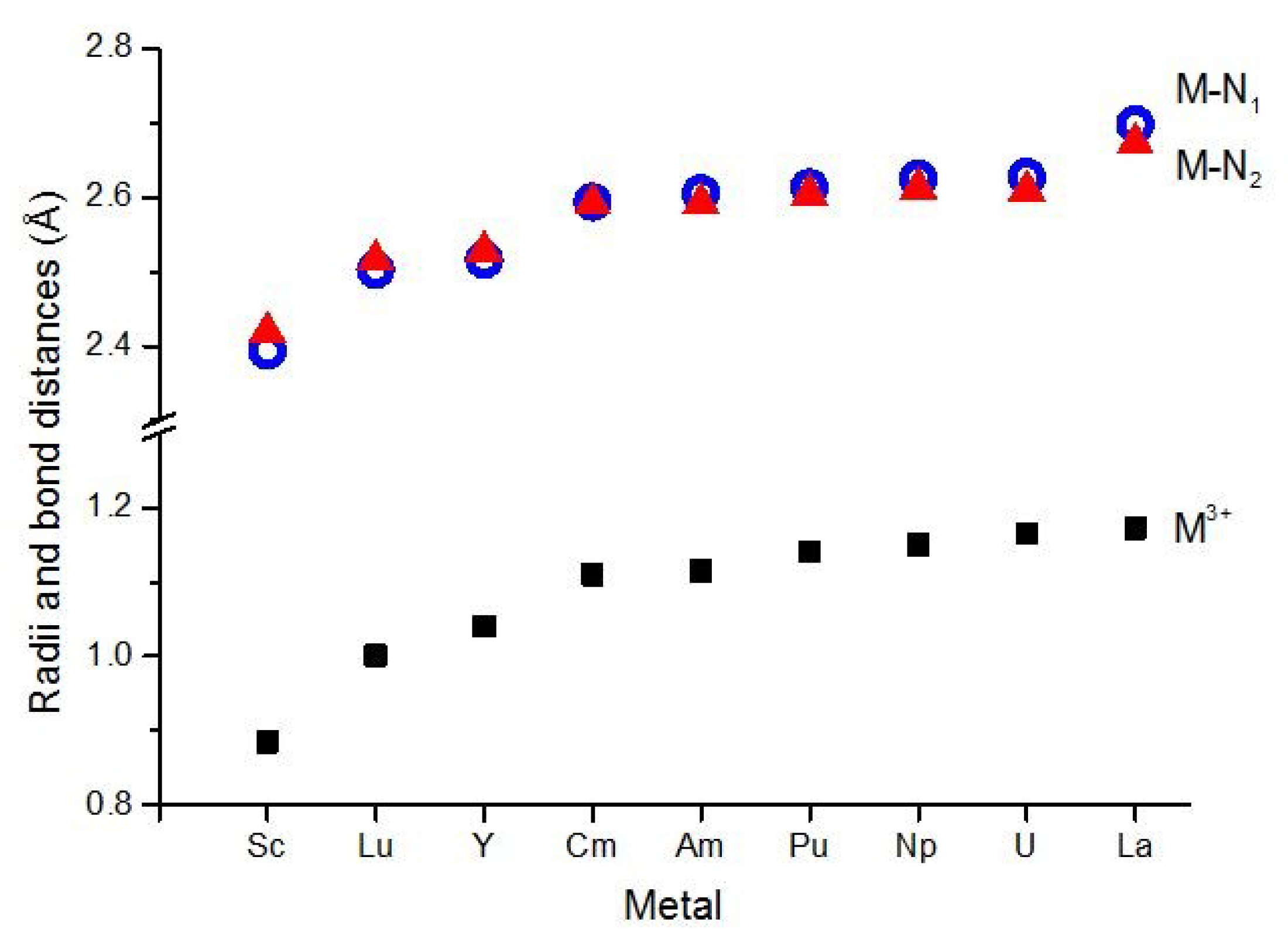
| M | r(III) b | Calculated | X-ray | |||||
|---|---|---|---|---|---|---|---|---|
| M–N1 | M–N2 | N1–C | N2–N3 | N2–C | M–N1 | M–N2 | ||
| Sc | 0.885 | 2.396 | 2.417 | 1.337 | 1.325 | 1.333 | 2.37 ± 0.01 | 2.41 ± 0.01 |
| Y | 1.040 | 2.516 | 2.525 | 1.339 | 1.328 | 1.334 | 2.49 ± 0.01 | 2.52 ± 0.02 |
| La | 1.172 | 2.684 | 2.658 | 1.342 | 1.328 | 1.333 | 2.611 ± 0.009 | 2.585 ± 0.009 |
| Lu | 1.001 | 2.500 | 2.509 | 1.339 | 1.328 | 1.334 | ||
| U | 1.165 | 2.627(9) | 2.609(10) | 1.342(1) | 1.329(1) | 1.335(1) | 2.55 ± 0.03 | 2.54 ± 0.03 |
| Np | 1.15 | 2.625(5) | 2.612(17) | 1.341(1) | 1.328(1) | 1.334(0) | ||
| Pu | 1.14 | 2.614(8) | 2.604(22) | 1.341(1) | 1.328(0) | 1.334(0) | 2.58 ± 0.02 | 2.55 ± 0.04 |
| Am | 1.115 | 2.605(3) | 2.592(20) | 1.341(2) | 1.328(1) | 1.333(2) | ||
| Cm | 1.11 | 2.594(1) | 2.592(4) | 1.340(0) | 1.328(1) | 1.334(0) | ||
| BTP′ | 1.337 | 1.330 | 1.344 | |||||
| Sc | Y | Pu | Am | Main Character b | ||||
|---|---|---|---|---|---|---|---|---|
| 1610, m | (1660) | 1606, m | (1657) | 1603, m | (1655) | 1603, m | (1656) | ν(Aza), ν(Py), ν(M–N) |
| 1585, w | (1644) | 1583, w | (1643) | 1580, w | (1642) | 1580, w | (1642) | ν(Aza), ν(Py), ν(M–N) |
| 1538, s | (1558) | 1534, sh | (1549) | 1532, s | (1545) | 1533, s | (1547) | ν(Aza), ν(Py), ν(M–N) |
| 1394, s | (1431) | 1390, s | (1428) | 1387, s | (1426) | 1388, s | (1427) | β(CH2), ν(Aza), ν(Py), ν(M–N) |
| 1018, m | (1045) | 1017, w | (1045) | 1014, m | (1043) | 1015, m | (1044) | β(Aza), ν(CC)propyl, ν(M–N) |
| 477, w | (455) | 470, w | (441) | 463, w | (437) | - | (438) | τ(Aza), γ(N–M) |
| 437, sh | (444) | hidden | (437) | 425, m | (435) | 425, m | (435) | τ(Aza), τ(Py), γ(N–M) |
| Property | Sc | Y | La | Lu | U | Np | Pu | Am | Cm |
|---|---|---|---|---|---|---|---|---|---|
| n(M) | +1.76 | +1.89 | +2.14 | +2.04 | +1.60 | +1.64 | +1.76 | +1.84 | +1.86 |
| n(N1) | −0.53 | −0.55 | −0.56 | −0.56 | −0.51 | −0.51 | −0.52 | −0.52 | −0.53 |
| n(N2) | −0.35 | −0.37 | −0.38 | −0.38 | −0.34 | −0.35 | −0.34 | −0.35 | −0.36 |
| s | 0.25 | 0.24 | 0.21 | 0.26 | 0.22 | 0.23 | 0.23 | 0.24 | 0.24 |
| d | 0.93 | 0.75 | 0.65 | 0.68 | 0.86 | 0.78 | 0.80 | 0.81 | 0.80 |
| f | - | - | - | - | 3.09 | 4.21 | 5.12 | 6.05 | 7.03 |
| N1(LP)→M | 218 | 194 | 140 | 164 | 436 | 269 | 264 | 252 | 219 |
| N2(LP)→M | 252 | 229 | 162 | 185 | 444 | 294 | 311 | 289 | 257 |
| N1–C→M | 56 | 69 | 43 | 59 | 79 | 65 | 68 | 63 | 41 |
| N2–N3→M | 39 | 58 | 32 | 46 | 62 | 55 | 55 | 53 | 24 |
| N2–C→M | 68 | 82 | 55 | 74 | 86 | 61 | 67 | 64 | 52 |
| M→Py | - | - | - | - | 7 | 10 | 18 | 8 | 5 |
| M→Aza | - | - | - | - | 8 | 13 | 24 | 9 | 4 |
| q(M) | +2.15 | +2.26 | +2.24 | +2.19 | +2.35 | +2.25 | +2.21 | +2.20 | +2.21 |
| ρ(M,N1) | 0.042 | 0.041 | 0.039 | 0.043 | 0.046 | 0.042 | 0.042 | 0.042 | 0.043 |
| ρ(M,N2) | 0.040 | 0.039 | 0.041 | 0.042 | 0.047 | 0.043 | 0.043 | 0.044 | 0.043 |
| ▽2ρ(M,N1) | 0.137 | 0.136 | 0.112 | 0.147 | 0.160 | 0.149 | 0.153 | 0.153 | 0.153 |
| ▽2ρ(M,N2) | 0.131 | 0.135 | 0.121 | 0.144 | 0.154 | 0.147 | 0.153 | 0.155 | 0.155 |
| Δ(M) | 0.99 | 0.97 | 1.11 | 0.98 | 1.50 | 1.29 | 1.26 | 1.22 | 1.17 |
| DI(M,N1) | 0.20 | 0.20 | 0.22 | 0.20 | 0.29 | 0.25 | 0.25 | 0.24 | 0.23 |
| DI(M,N2) | 0.19 | 0.19 | 0.22 | 0.19 | 0.28 | 0.25 | 0.25 | 0.24 | 0.23 |
| De b | 3406 | 3019 | 2645 | 3058 | 2735 | 2746 | 2815 | 2842 | 2851 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, A.; Apostolidis, C.; Walter, O. Comparative Study of Complexes of Rare Earths and Actinides with 2,6-Bis(1,2,4-triazin-3-yl)pyridine. Inorganics 2019, 7, 26. https://doi.org/10.3390/inorganics7030026
Kovács A, Apostolidis C, Walter O. Comparative Study of Complexes of Rare Earths and Actinides with 2,6-Bis(1,2,4-triazin-3-yl)pyridine. Inorganics. 2019; 7(3):26. https://doi.org/10.3390/inorganics7030026
Chicago/Turabian StyleKovács, Attila, Christos Apostolidis, and Olaf Walter. 2019. "Comparative Study of Complexes of Rare Earths and Actinides with 2,6-Bis(1,2,4-triazin-3-yl)pyridine" Inorganics 7, no. 3: 26. https://doi.org/10.3390/inorganics7030026
APA StyleKovács, A., Apostolidis, C., & Walter, O. (2019). Comparative Study of Complexes of Rare Earths and Actinides with 2,6-Bis(1,2,4-triazin-3-yl)pyridine. Inorganics, 7(3), 26. https://doi.org/10.3390/inorganics7030026