Tuning the Linear and Nonlinear Optical Properties of Pyrene-Pyridine Chromophores by Protonation and Complexation to d10 Metal Centers §

 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
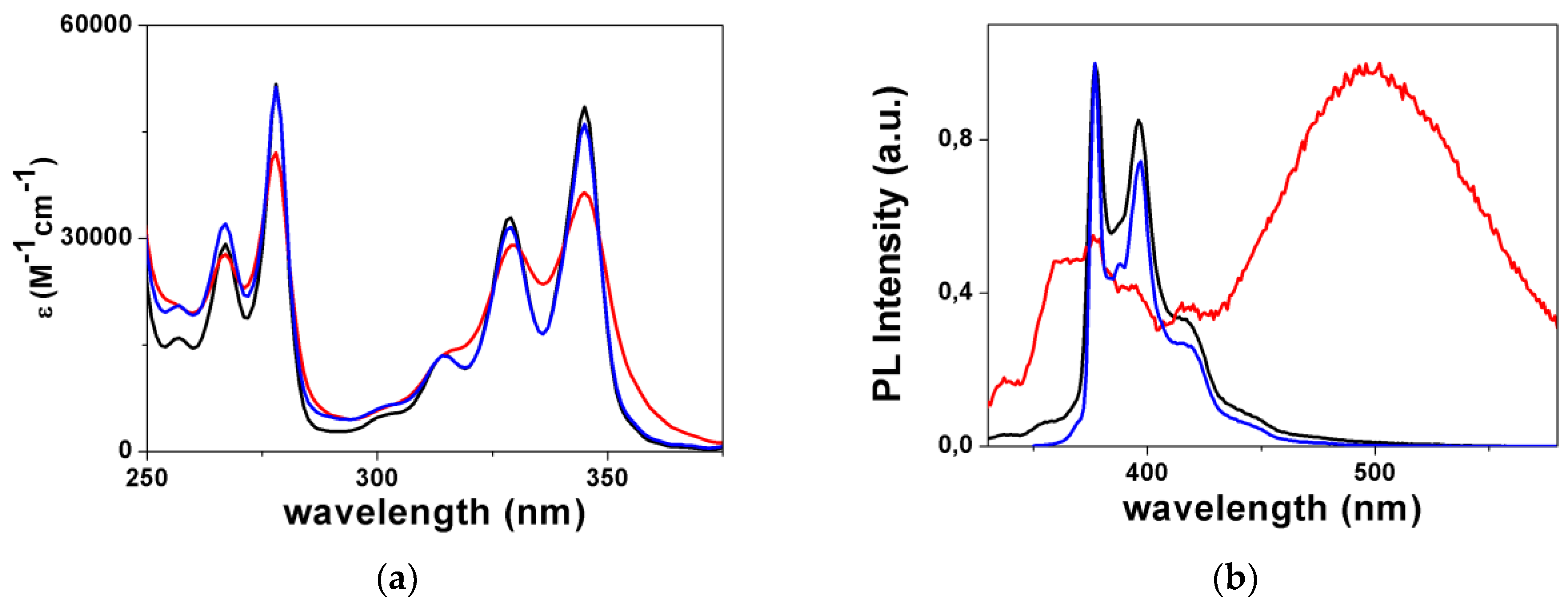
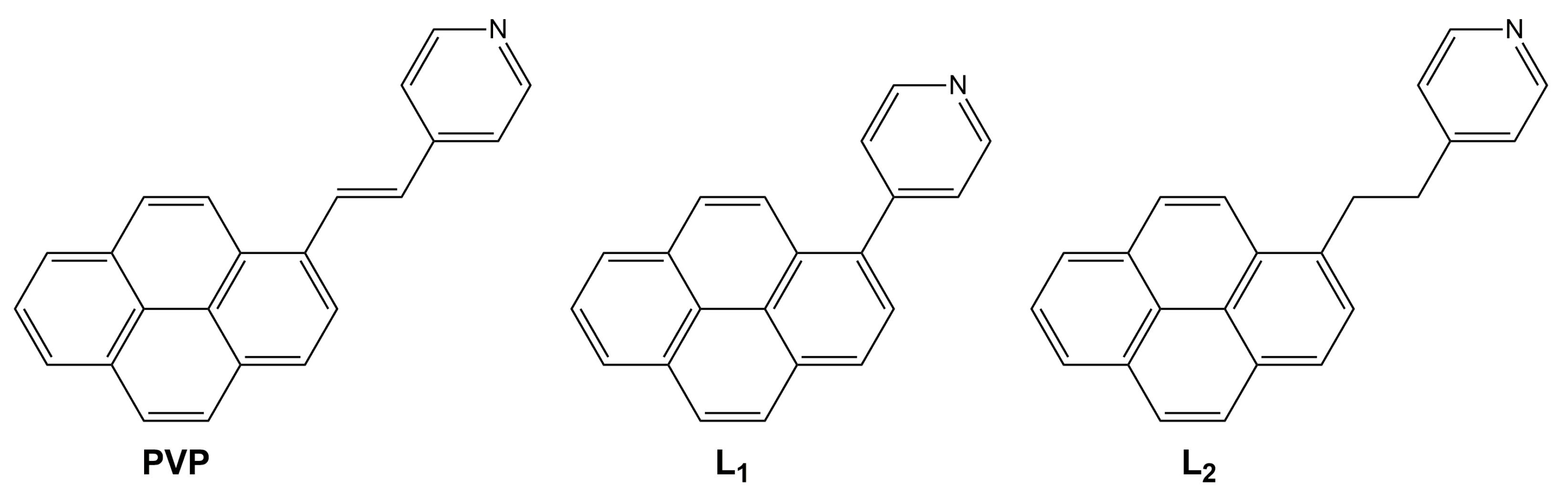
2.1. Synthesis and Characterization of L1, [L1H]+, and ZnL1
2.2. Synthesis and Characterization of L2, [L2H]+, and CuL2
3. Materials and Methods
3.1. Single-Crystal X-ray Crystallographic Studies
3.2. Computational Details
3.3. EFISH Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Papadopoulos, M.G.; Sadlej, A.J.; Leszczynski, J. Non-Linear Optical Properties of Matter; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Zyss, J. Molecular Nonlinear Optics: Materials, Physics, and Devices; Academic Press: Boston, MA, USA, 1994. [Google Scholar]
- Lesley, M.J.G.; Woodward, A.; Taylor, N.J.; Marder, T.B.; Cazenobe, I.; Ledoux, I.; Zyss, J.; Thornton, A.; Bruce, D.W.; Kakkar, A.K. Lewis Acidic Borane Adducts of Pyridines and Stilbazoles for Nonlinear Optics. Chem. Mater. 1998, 10, 1355–1365. [Google Scholar] [CrossRef]
- Evans, O.R.; Lin, W. Crystal Engineering of NLO Materials Based on Metal–Organic Coordination Networks. Acc. Chem. Res. 2002, 35, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Cariati, E.; Pizzotti, M.; Roberto, D.; Tessore, F.; Ugo, R. Coordination and organometallic compounds and inorganic–organic hybrid crystalline materials for second-order non-linear optics. Coord. Chem. Rev. 2006, 250, 1210–1233. [Google Scholar] [CrossRef]
- Kanis, D.R.; Lacroix, P.G.; Ratner, M.A.; Marks, T.J. Electronic Structure and Quadratic Hyperpolarizabilities in Organotransition-Metal Chromophores Having Weakly Coupled π-Networks. Unusual Mechanisms for Second-Order Response. J. Am. Chem. Soc. 1994, 116, 10089–10102. [Google Scholar] [CrossRef]
- Roberto, D.; Ugo, R.; Bruni, S.; Cariati, E.; Cariati, F.; Fantucci, P.; Invernizzi, I.; Quici, S.; Ledoux, I.; Zyss, J. Quadratic Hyperpolarizability Enhancement of para-Substituted Pyridines upon Coordination to Organometallic Moieties: The Ambivalent Donor or Acceptor Role of the Metal. Organometallics 2000, 19, 1775–1788. [Google Scholar] [CrossRef]
- Cariati, E.; Botta, C.; Danelli, S.G.; Forni, A.; Giaretta, A.; Giovanella, U.; Lucenti, E.; Marinotto, D.; Righetto, S.; Ugo, R. Solid state and solution fine tuning of the linear and nonlinear optical properties of (2-pyrene-1-yl-vinyl)pyridine by protonation-deprotonation reactions. Chem. Commun. 2014, 50, 14225–14228. [Google Scholar] [CrossRef] [PubMed]
- Cariati, E.; Dragonetti, C.; Lucenti, E.; Nisic, F.; Righetto, S.; Roberto, D.; Tordin, E. An acido-triggered reversible luminescent and nonlinear optical switch based on a substituted styrylpyridine: EFISH measurements as an unusual method to reveal a protonation-deprotonation NLO contrast. Chem. Commun. 2014, 50, 1608. [Google Scholar] [CrossRef] [PubMed]
- Creus, J.; Matheu, R.; Peñafiel, I.; Moonshiram, D.; Blondeau, P.; Benet-Buchholz, J.; García-Antón, J.; Sala, X.; Godard, C.; Llobet, A. A Million Turnover Molecular Anode for Catalytic Water Oxidation. Angew. Chem. Int. Ed. 2016, 55, 15382–15386. [Google Scholar] [CrossRef] [Green Version]
- Hüglin, D.; Seiffert, W.; Zimmermann, H.W. Time-resolved microfluorometric study of the binding sites of lipophilic cationic pyrene probes in mitochondria of living HeLa cells. J. Photochem. Photobiol. B Biol. 1995, 31, 145–158. [Google Scholar] [CrossRef]
- Sankaran, N.B.; Banthia, S.; Das, A.; Samanta, A. Fluorescence signaling of transition metal ions: A new approach. New J. Chem. 2002, 26, 1529–1531. [Google Scholar] [CrossRef]
- Sankaran, N.B.; Das, A.; Samanta, A. Interaction between a pyridyl and a naphthyl/pyrenyl moiety in covalently linked systems. Chem. Phys. Lett. 2002, 351, 61–70. [Google Scholar] [CrossRef]
- Camerman, A.; Trotter, J. The crystal and molecular structure of pyrene. Acta Cryst. 1965, 18, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Pielak, K.; Tonnelé, C.; Sanguinet, L.; Cariati, E.; Righetto, S.; Muccioli, L.; Castet, F.; Champagne, B. Dynamical Behavior and Second Harmonic Generation Responses in Acido-Triggered Molecular Switches. J. Phys. Chem. 2018, 122, 26160–26168. [Google Scholar] [CrossRef]
- Dojer, B.; Pevec, A.; Belaj, F.; Kristl, M. Two new zinc(II) acetates with 3- and 4-aminopyridine: Syntheses and structural properties. Acta Chim. Slov. 2015, 62, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Roberto, D.; Ugo, R.; Tessore, F.; Lucenti, E.; Quici, S.; Vezza, S.; Fantucci, P.; Invernizzi, I.; Bruni, S.; Ledoux-Rak, I.; et al. Effect of the Coordination to M(II) Metal Centers (M = Zn, Cd, Pt) on the Quadratic Hyperpolarizability of Various Substituted 5-X-1,10-phenanthrolines (X = Donor Group) and of trans-4-(Dimethylamino)-4′-stilbazole. Organometallics 2002, 21, 161–170. [Google Scholar] [CrossRef]
- Di Bella, S.; Colombo, A.; Dragonetti, C.; Righetto, S.; Roberto, D. Zinc(II) as a Versatile Template for Efficient Dipolar and Octupolar Second-Order Nonlinear Optical Molecular Materials. Inorganics 2018, 6, 133. [Google Scholar] [CrossRef]
- Kollár, J.; Hrdlovič, P.; Chmela, Š. Spectral properties of bichromophoric pyrene derivatives: Monomer vs. excimer fluorescence. J. Photochem. Photobiol. Chem. 2010, 214, 33–39. [Google Scholar] [CrossRef]
- Filby, M.H.; Dickson, S.J.; Zaccheroni, N.; Prodi, L.; Bonacchi, S.; Montalti, M.; Paterson, M.J.; Humphries, T.D.; Chiorboli, C.; Steed, J.W. Induced Fit Interanion Discrimination by Binding-Induced Excimer Formation. J. Am. Chem. Soc. 2008, 130, 4105–4113. [Google Scholar] [CrossRef]
- Cariati, E.; Roberto, D.; Ugo, R.; Ford, P.C.; Galli, S.; Sironi, A. New Structural Motifs, Unusual Quenching of the Emission, and Second Harmonic Generation of Copper(I) Iodide Polymeric or Oligomeric Adducts with Para-Substituted Pyridines or trans-Stilbazoles. Inorg. Chem. 2005, 44, 4077–4085. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.X.; et al. Gaussian 16, Revision A.03; Gaussian: Wallingford, CT, USA, 2016. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Johnson, L.E.; Dalton, L.R.; Robinson, B.H. Optimizing Calculations of Electronic Excitations and Relative Hyperpolarizabilities of Electrooptic Chromophores. Acc. Chem. Res. 2014, 47, 3258–3265. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, I.; Zyss, J. Influence of the molecular environment in solution measurements of the Second-order optical susceptibility for urea and derivatives. Chem. Phys. 1982, 73, 203–213. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λabs (nm) | λem a (nm) | μβλ (10−48 esu) |
|---|---|---|---|
| Pyrene | 337, 322, 309, 274, 264 | 415, 394, 385, 374 b | - |
| PVP c | 378(br) | 450 | 1500 |
| [PVPH]+ c | 443, 395 | 550 | −950 |
| L1 | 345, 330(sh), 314(sh), 280, 270(sh) | 407, 390 | 650 ± 156 |
| [L1H]+ | 410, 380, 328, 304 | 482 | −460 ± 78 |
| ZnL1 | 345, 330(sh), 314(sh), 280, 270(sh) | 407, 390 | −250 ± 42 |
| L2 | 345, 328, 314, 277, 266 | 440(sh), 415(sh), 396, 377 | 197 ± 73 |
| [L2H]+ | 345, 328, 314, 277, 266 | 496 | −270 ± 62 |
| CuL2 | 345, 328, 314, 277, 266 | 443(sh), 416(sh), 397, 377 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucenti, E.; Forni, A.; Marinotto, D.; Previtali, A.; Righetto, S.; Cariati, E. Tuning the Linear and Nonlinear Optical Properties of Pyrene-Pyridine Chromophores by Protonation and Complexation to d10 Metal Centers §. Inorganics 2019, 7, 38. https://doi.org/10.3390/inorganics7030038
Lucenti E, Forni A, Marinotto D, Previtali A, Righetto S, Cariati E. Tuning the Linear and Nonlinear Optical Properties of Pyrene-Pyridine Chromophores by Protonation and Complexation to d10 Metal Centers §. Inorganics. 2019; 7(3):38. https://doi.org/10.3390/inorganics7030038
Chicago/Turabian StyleLucenti, Elena, Alessandra Forni, Daniele Marinotto, Andrea Previtali, Stefania Righetto, and Elena Cariati. 2019. "Tuning the Linear and Nonlinear Optical Properties of Pyrene-Pyridine Chromophores by Protonation and Complexation to d10 Metal Centers §" Inorganics 7, no. 3: 38. https://doi.org/10.3390/inorganics7030038
APA StyleLucenti, E., Forni, A., Marinotto, D., Previtali, A., Righetto, S., & Cariati, E. (2019). Tuning the Linear and Nonlinear Optical Properties of Pyrene-Pyridine Chromophores by Protonation and Complexation to d10 Metal Centers §. Inorganics, 7(3), 38. https://doi.org/10.3390/inorganics7030038