Water Network Dynamics Next to the Oxygen-Evolving Complex of Photosystem II


Abstract
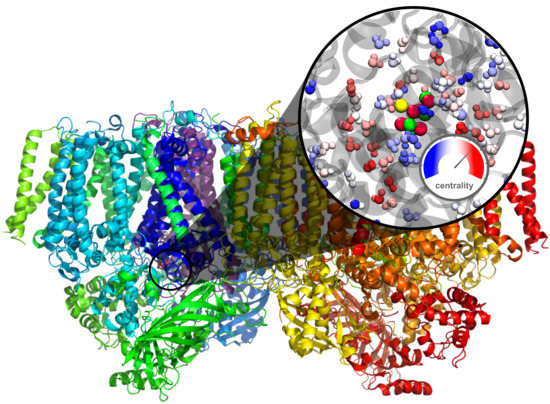
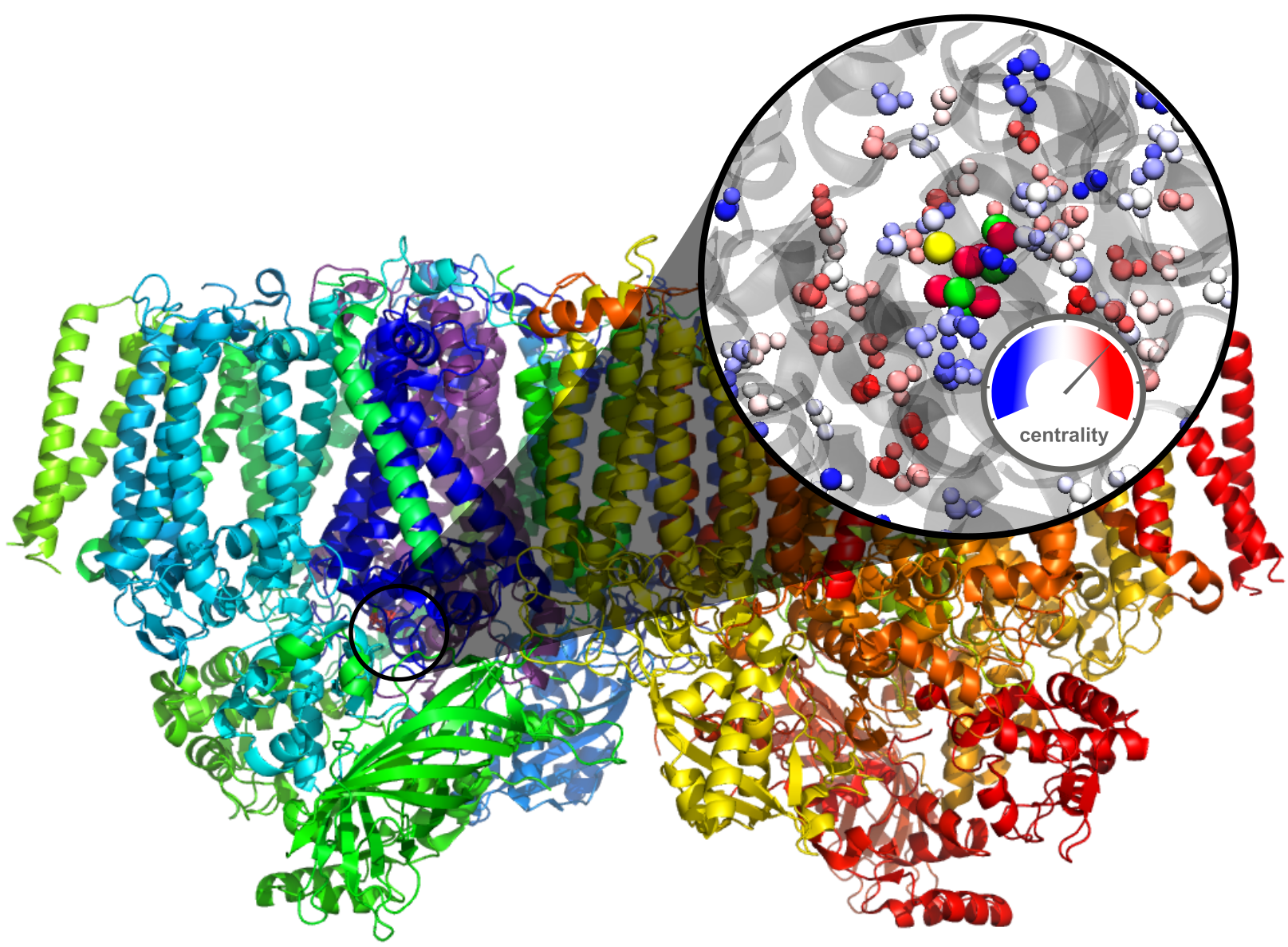
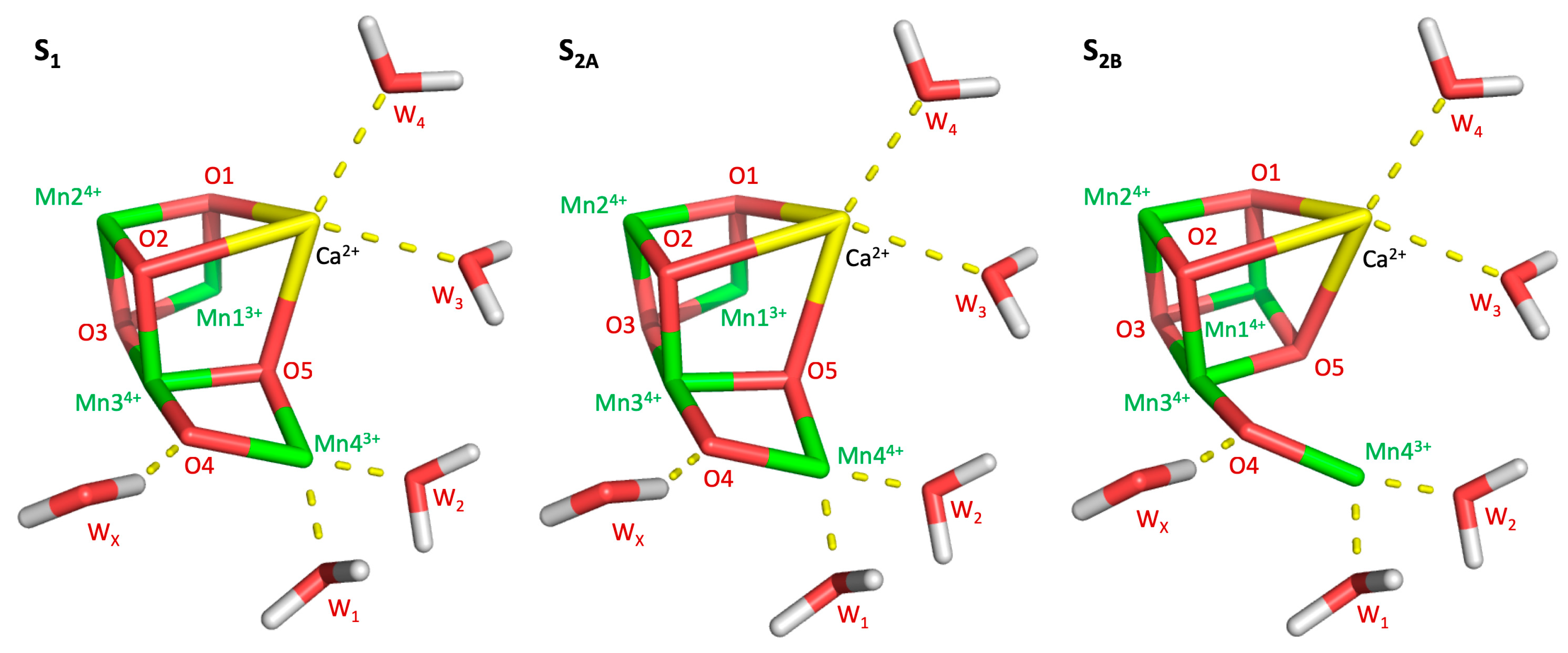
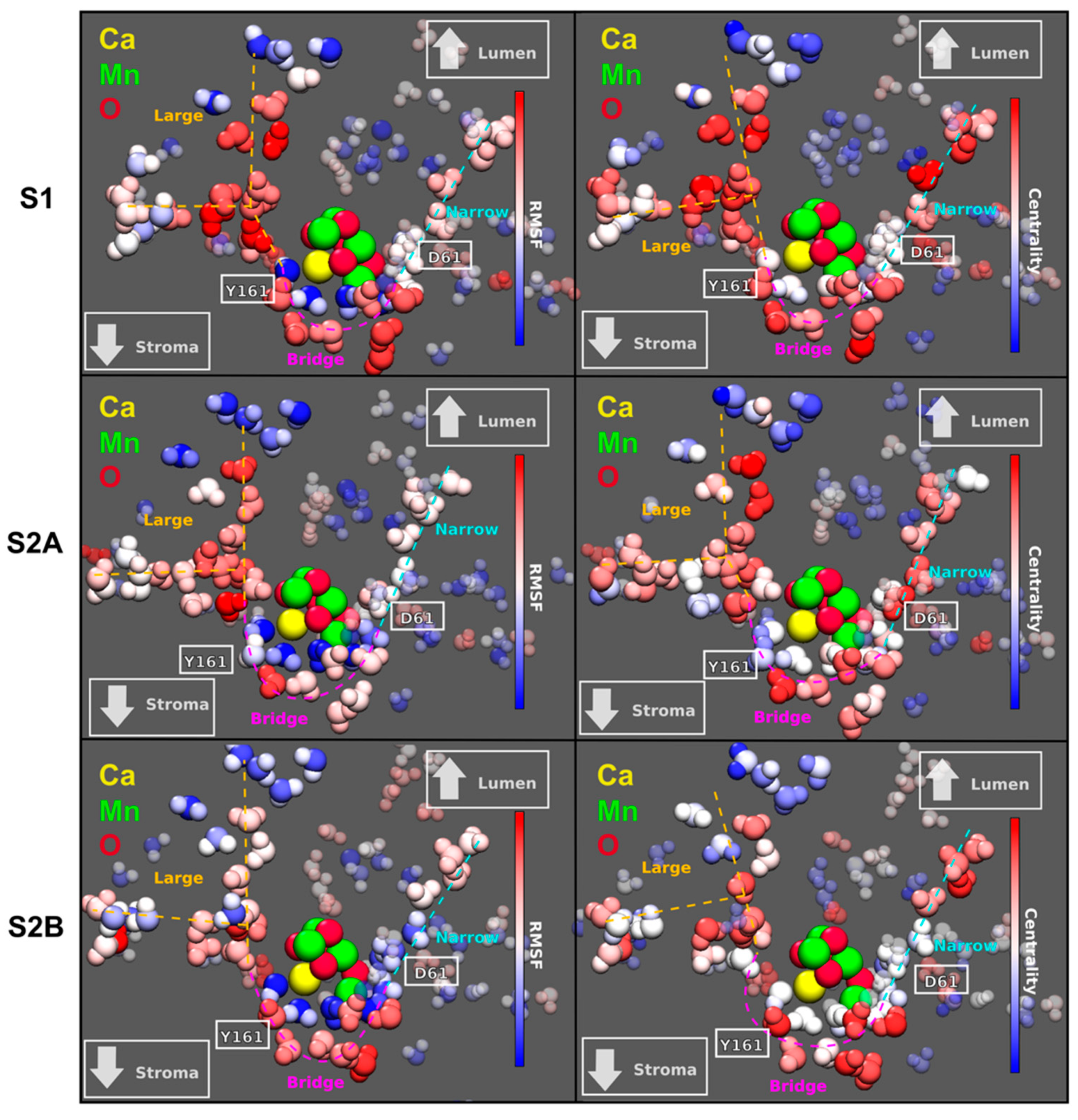
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
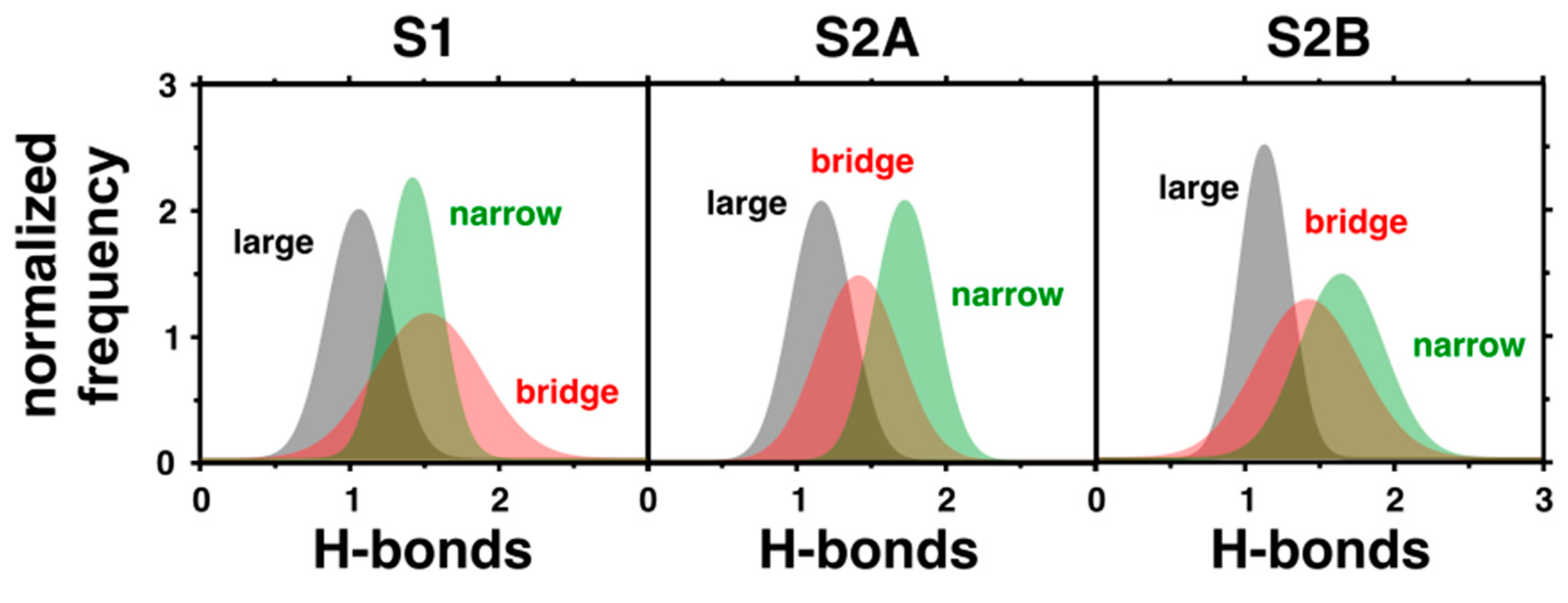
2.1. Water Network Dynamics
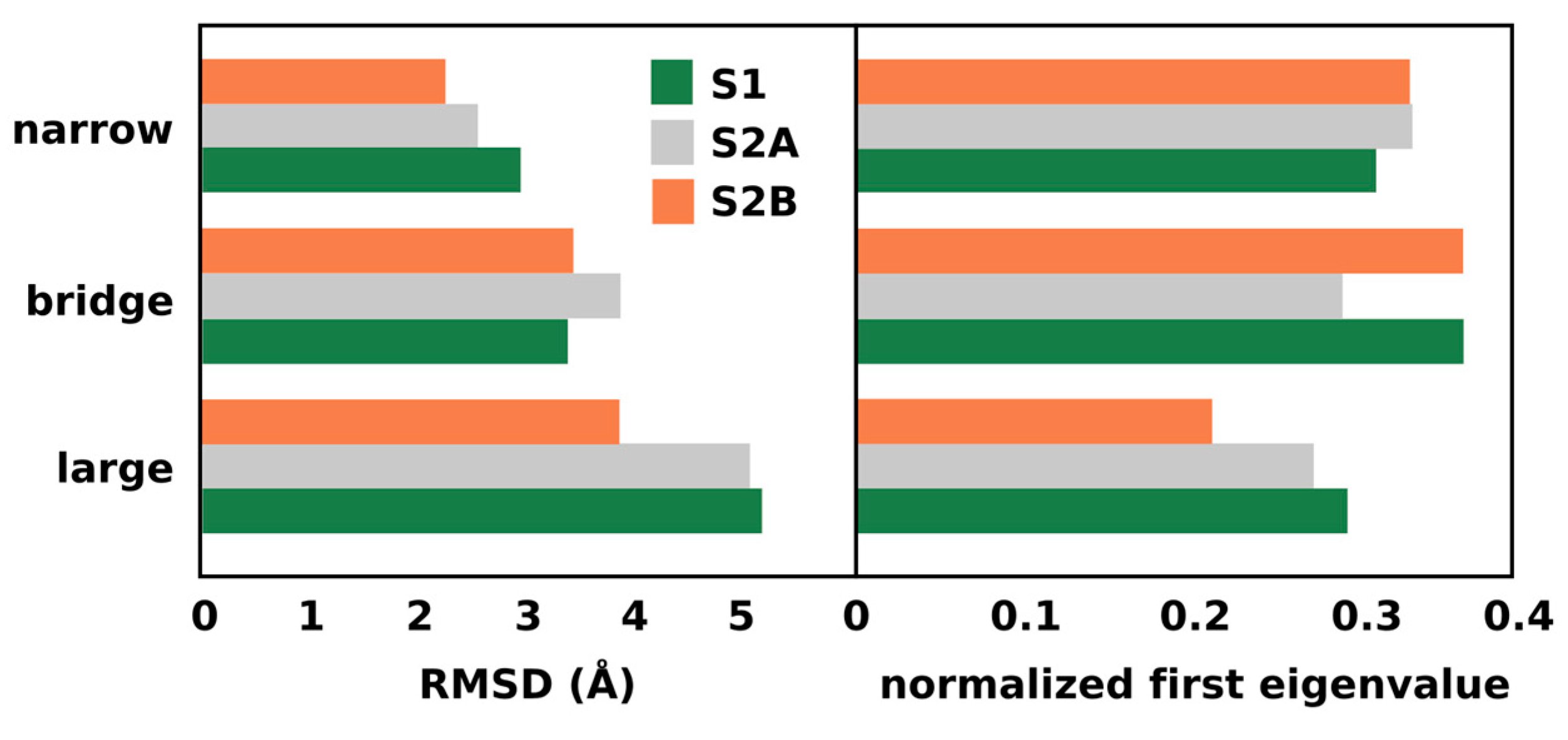
2.2. Diffusion vs. Connectivity
3. Discussion
4. Materials and Methods
4.1. MD Simulations
4.2. Eigenvector Centrality Distribution for Water Networks
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kok, B.; Forbush, B.; McGloin, M. Cooperation of charges in photosynthetic oxygen evolution—I. A linear four step mechanism. Photochem. Photobiol. 1970, 11, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Joliot, P.; Barbieri, G.; Chabaud, R. Model of the system II photochemical centers. Photochem. Photobiol. 1969, 10, 309–329. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Ames, W.; Cox, N.; Lubitz, W.; Neese, F. Two interconvertible structures that explain the spectroscopic properties of the oxygen-evolving complex of photosystem II in the S2 state. Angew. Chem. Int. 2012, 51, 9935–9940. [Google Scholar] [CrossRef] [PubMed]
- Dismukes, G.C.; Siderer, Y. Intermediates of a polynuclear manganese center involved in photosynthetic oxidation of water. Proc. Natl. Acad. Sci. USA 1981, 78, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, J.L.; Sauer, K. EPR detection of a cryogenically photogenerated intermediate in photosynthetic oxygen evolution. Biochim. Biophys. Acta 1984, 767, 21–28. [Google Scholar] [CrossRef]
- Zimmermann, J.L.; Rutherford, A.W. Electron paramagnetic resonance properties of the S2 state of the oxygen-evolving complex of photosystem II. Biochemistry 1986, 25, 4609–4615. [Google Scholar] [CrossRef]
- Askerka, M.; Wang, J.; Vinyard, D.J.; Brudvig, G.W.; Batista, V.S. S3 state of the O2-evolving complex of photosystem II: Insights from QM/MM, EXAFS, and femtosecond X-ray diffraction. Biochemistry 2016, 55, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Vass, I.; Styring, S. pH-dependent charge equilibria between tyrosine-D and the S states in photosystem II: Estimation of relative midpoint redox potentials. Biochemistry 1991, 30, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Askerka, M.; Wang, J.; Brudvig, G.W.; Batista, V.S. Structural changes in the oxygen-evolving complex of photosystem II induced by the S1 to S2 transition: A combined XRD and QM/MM study. Biochemistry 2014, 53, 6860–6862. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Ota, K.; Shibuya, Y.; Noguchi, T. Role of a water network around the Mn4CaO5 cluster in photosynthetic water oxidation: A Fourier transform infrared spectroscopy and quantum mechanics/molecular mechanics calculation study. Biochemistry 2016, 55, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Retegan, M.; Pantazis, D.A. Differences in the active site of water oxidation among photosynthetic organisms. J. Am. Chem. Soc. 2017, 139, 14340–14343. [Google Scholar] [CrossRef] [PubMed]
- Vassiliev, S.; Zaraiskaya, T.; Bruce, D. Molecular dynamics simulations reveal highly permeable oxygen exit channels shared with water uptake channels in photosystem II. Biochim. Biophys. Acta 2013, 1827, 1148–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakashita, N.; Watanabe, H.C.; Ikeda, T.; Saito, K.; Ishikita, H. Origins of water molecules in the photosystem II crystal structure. Biochemistry 2017, 56, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Vassiliev, S.; Zaraiskaya, T.; Bruce, D. Exploring the energetics of water permeation in photosystem II by multiple steered molecular dynamics simulations. Biochim. Biophys. Acta 2012, 1817, 1671–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, K.; Yuki, T.; Hatakeyama, M.; Uchida, W.; Nakamura, S. All-atom molecular dynamics simulation of photosystem II embedded in thylakoid membrane. J. Am. Chem. Soc. 2013, 135, 15670–15673. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Askerka, M.; Brudvig, G.W.; Batista, V.S. Crystallographic data support the carousel mechanism of water supply to the oxygen-evolving complex of photosystem II. ACS Energy Lett. 2017, 2, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Ugur, I.; Rutherford, A.W.; Kaila, V.R. Redox-coupled substrate water reorganization in the active site of photosystem II—The role of calcium in substrate water delivery. Biochim. Biophys. Acta Bioenergy 2016, 1857, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Retegan, M.; Pantazis, D.A. Interaction of methanol with the oxygen-evolving complex: Atomistic models, channel identification, species dependence, and mechanistic implications. Chem. Sci. 2016, 7, 6463–6476. [Google Scholar] [CrossRef] [PubMed]
- Oyala, P.H.; Stich, T.A.; Debus, R.J.; Britt, R.D. Ammonia binds to the dangler manganese of the photosystem II oxygen-evolving complex. J. Am. Chem. Soc. 2015, 137, 8829–8837. [Google Scholar] [CrossRef] [PubMed]
- Askerka, M.; Vinyard, D.J.; Brudvig, G.W.; Batista, V.S. NH3 binding to the S2 state of the O2-evolving complex of photosystem II: Analogue to H2O binding during the S2→S3 transition. Biochemistry 2015, 54, 5783–5786. [Google Scholar] [CrossRef] [PubMed]
- Kraskov, A.; Stögbauer, H.; Grassberger, P. Estimating mutual information. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2004, 69, 066138. [Google Scholar] [CrossRef] [PubMed]
- Askerka, M.; Brudvig, G.W.; Batista, V.S. The O2-evoling complex of photosystem II: Recent insights from quantum mechanics/molecular mechanics (QM/MM), extended X-ray absorption fine structure (EXAFS), and femtosecond x-ray crystallography data. Acc. Chem. Res. 2017, 50, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ishikita, H.; Saenger, W.; Loll, B.; Biesaidka, J.; Knapp, E.W. Energetics of a possible proton exit pathway for water oxidation in photosystem II. Biochemistry 2006, 45, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Batista, V.S.; Brudvig, G.W. Occupancy of water molecules near the oxygen-evolving complex of photosystem II. Protein Sci. 2019. under review. [Google Scholar]
- Umena, Y.; Kawakami, K.; Shen, J.R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at resolution 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Wiwczar, J.M.; LaFountain, A.M.; Wang, J.M.; Frank, H.A.; Brudvig, G.W. Chlorophyll a with a farnesyl tail in thermophilic cyanobacteria. Photosyn. Res. 2017, 134, 175–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wincencjusz, H.; vanGorkom, H.J.; Yocum, C.F. The Photosynthetic Oxygen Evolving Complex Requires Chloride for its redox states S2→S3 and S3→S0 transition but not for S0→S1 or S1→S2 transitions. Biochemistry 1997, 36, 3663–3670. [Google Scholar] [CrossRef] [PubMed]
- Rivalta, I.; Amin, M.; Luber, S.; Vassiliev, S.; Pokhrel, R.; Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N.; Bruce, D.; et al. Structural/functional role of chloride in photosystem II. Biochemistry 2011, 50, 6312–6315. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, R.; Service, R.J.; Debus, R.J.; Brudvig, G.W. Mutation of lysine 317 in the D2 subunit of photosystem II alters chloride binding and proton transport. Biochemistry 2013, 52, 4758–4773. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comp. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian09 Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations potentials for the transition-metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–285. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of polarization functions on molecular-orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Seminario, J.M. Calculation of intramolecular force fields from second-derivative tensors. Int. J. Quantum Chem. 1996, 60, 1271–1277. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Peters, M.B.; Yang, Y.; Wang, B.; Fusti-Molnar, L.; Weaver, M.N.; Merz, K.M. Structural survey of zinc-containing proteins and development of the Zinc Amber Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Merz, K.M. MCPB.py: A Python based metal center parameter builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AmberTools18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Negre, C.F.A.; Morzan, U.N.; Hendrickson, H.P.; Pal, R.; Lisi, G.P.; Loria, J.P.; Rivalta, I.; Ho, J.; Batista, V.S. Eigenvector centrality for characterization of protein allosteric pathways. Proc. Natl. Acad. Sci. USA 2018, 115, 12201–12208. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bells Lab Tech. 1948, 27, 623–656. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiss, K.; Morzan, U.N.; Grigas, A.T.; Batista, V.S. Water Network Dynamics Next to the Oxygen-Evolving Complex of Photosystem II. Inorganics 2019, 7, 39. https://doi.org/10.3390/inorganics7030039
Reiss K, Morzan UN, Grigas AT, Batista VS. Water Network Dynamics Next to the Oxygen-Evolving Complex of Photosystem II. Inorganics. 2019; 7(3):39. https://doi.org/10.3390/inorganics7030039
Chicago/Turabian StyleReiss, Krystle, Uriel N. Morzan, Alex T. Grigas, and Victor S. Batista. 2019. "Water Network Dynamics Next to the Oxygen-Evolving Complex of Photosystem II" Inorganics 7, no. 3: 39. https://doi.org/10.3390/inorganics7030039
APA StyleReiss, K., Morzan, U. N., Grigas, A. T., & Batista, V. S. (2019). Water Network Dynamics Next to the Oxygen-Evolving Complex of Photosystem II. Inorganics, 7(3), 39. https://doi.org/10.3390/inorganics7030039